

Subscribe to RSS
DOI: 10.1055/s-0037-1610382
Recent Developments in Polyene Cyclizations and Their Applications in Natural Product Synthesis
Authors
Publication History
Received: 15 October 2018
Accepted: 18 October 2018
Publication Date:
15 November 2018 (online)

Published as part of the 50 Years SYNTHESIS – Golden Anniversary Issue
Abstract
Cascade polyene cyclization reactions are highly efficient and elegant bioinspired transformations that involve simultaneous multiple bond constructions to rapidly generate complex polycyclic molecules. This review summarizes the most prominent work on a variety of cationic and radical cascade cyclizations and their applications in natural product synthesis published between 2014 and 2018.
1 Introduction
2 Cationic Polyene Cyclizations
2.1 Lewis Acid Mediated Polyene Cyclizations
2.2 Brønsted Acid Mediated Polyene Cyclizations
2.3 Halogen Electrophile Initiated Polyene Cyclizations
2.4 Sulfur Electrophile Initiated Polyene Cyclizations
2.5 Transition-Metal-Mediated Cationic Polyene Cyclizations
3 Radical Polyene Cyclizations
3.1 Transition-Metal-Mediated Radical Polyene Cyclizations
3.2 Photocatalyst-Mediated Polyene Cyclizations
4 Origin of Stereocontrol in Polyene Cyclizations
5 Conclusion
Key words
polyene cyclization - cascade reaction - carbon–carbon bond formation - natural products - total synthesisIntroduction
Biomimetic strategies are powerful methods in natural product synthesis since they can provide concise methods for the elaboration of structurally complex molecules from significantly simpler precursors. Polyene cyclizations are recognized as unique and efficient ways to build structural complexity from acyclic molecules via concerted and stereocontrolled C–C bond formations. In 1955, Stork and Eschenmoser independently published a hypothesis rationalizing the stereoelectronic aspects that underpin the stereochemical outcomes of polyene cyclizations, which immediately encouraged further studies on these types of transformations.[1] In 1968 and 1975, pioneering research on the biomimetic syntheses of steroids was reported by Johnson[2] and van Tamelen,[3] both of whom demonstrated the profound synthetic power of polyene cyclizations in natural product total synthesis.

Inspired by these revolutionary investigations into the use of polyene cyclizations in organic synthesis, considerable advancements have been achieved in the past several decades to improve the efficiency of this biomimetic process, develop novel creative variants of the cyclization processes, as well as invent novel chiral reagents and catalysts to induce highly enantioselective and regioselective cyclizations, with comparable selectivity to the enzyme-controlled cyclizations in nature.[4] Snyder, Gagné, Anderson, and Sarlah previously reviewed this topic from 2000 to 2016.[4] The aim of this review is to provide an updated overview of cascade polyene cyclizations and their applications toward natural product synthesis from 2014 to 2018. These cyclization processes are first divided mechanistically into cationic and radical pathways and are further subcategorized according to the mode of activation.
2
Cationic Polyene Cyclizations
2.1Lewis Acid Mediated Polyene Cyclizations
In 2014, Li and co-workers reported the synthesis of antitumor and anti-inflammatory natural products triptolide (4), triptonide (5), and related analogues which utilized a series of metal-mediated reactions (Scheme [1]).[5] Functionalization of carboxylic acid 1 provided alkyne 2. The subsequent key step involved an indium bromide mediated polyene cyclization, which was originally developed by Corey and co-workers,[6] to furnish the trans-fused ring framework 3 (75%).


The Barrett group has applied a dual biomimetic approach to the synthesis of (+)-hongoquercin B (9) by successive polyketide and polyene cascade cyclizations (Scheme [2]).[7] Cyclo-aromatization of β,δ-diketo-dioxinone 6 and subsequent enantioselective epoxidation provided resorcylate 7. Reaction of resorcylate 7 with iron(III) chloride hexahydrate catalyzed polyene cyclization to provide the meroterpenoid 8 (56%, 92% ee) as a single diastereomer and demonstrated that the single epoxide stereocenter controlled the resulting five stereocenters formed during this transformation.
In the synthesis of atropisomeric indoloterpenoid natural product (+)-dixiamycin B (12), the Baran group utilized a boron trifluoride–diethyl ether complex mediated cyclization of epoxide 10 into alcohol 11 as the key step to furnish the pentacyclic core (Scheme [3]).[8] It was observed that the N-protection with Boc2O was essential in order to suppress the formation of undesired isomers during polyene cyclization, thereby enhancing the yield of alcohol 11.

While polyene cyclization reactions are often used in the synthesis of fused polycyclic ring systems, their use in the construction of bridged polycyclic ring systems was only recently, in 2015, explored by Nakamura and co-workers (Scheme [4]).[9] Exposure of epoxyallylsilane 13 to diethylaluminum chloride resulted in polyene cyclization via carbocation intermediates 14 and 15 to produce bridged tricyclic compound 16 (72%) after hydrolysis. Tetracyclic product 18 (37%) was also obtained under the same conditions from epoxyallylsilane 17.

A systematic study of using functionalized unsaturated oxiranes in Lewis acid mediated polyene cyclization was published by Corey and co-workers, providing valuable insight into the electronic effects of neighboring substituents in epoxide initiated cationic π-cyclizations (Scheme [5]).[10] Enantiomerically enriched epoxy alcohol 21 was efficiently prepared from alcohol 19 via enantioselective Katsuki–Sharpless epoxidation to give epoxide 20 (86%, 90% ee). Further derivatization of epoxy alcohol 21 gave functionalized oxiranes 22a–26a. It was found that aldehyde 22a (Scheme [5b], entry 1) and ester 23a (Scheme [5b], entry 2) were unreactive when exposed to tin(IV) chloride. It is postulated that chelation of tin(IV) chloride by the epoxide and carbonyl groups did not facilitate cyclization, but prevent it from occurring as the carbonyl group becomes electron deficient, inhibiting the heterolysis of the oxirane α-C–O bond. In contrast, reaction of methoxime 24a (Scheme [5b], entry 3) with tin(IV) chloride resulted in polyene cyclization to give tricyclic product 24b (95%), which could be explained by the electron-donating nature of the methoxy group, favoring the heterolytic fission of the oxirane α-C–O bond. Vinyloxirane 25a (Scheme [5b], entry 4) was also smoothly cyclized to provide the tricyclic product 25b (84%) on reaction with tin(IV) chloride. This result demonstrates that substitution on the epoxide may facilitate the Lewis acid activation of the epoxide moiety; the resulting polyene cyclization is assisted by the π-electron-donating nature of its substituent. Furthermore, it was found that conversion of epoxide alcohol 21 into alkoxytrichlorostannane complex 26a (Scheme [5b], entry 5) also facilitated epoxide activation by covalently attaching a Lewis acid to the substrate, thereby leading to tricyclic diol 26b (82%) on destannylative work-up.

Celastroid natural products have a range of promising biological activities and are being studied in clinical trials. Seigel and co-workers reported the first total synthesis of celastrol (29) which utilized polyene cyclization as the key step to elaborate the pentacyclic core (Scheme [6]).[11] Iron(III) chloride catalyzed polyene cyclization of allylic alcohol 27 produced pentacyclic alkene 28 (38%) on a gram scale. The use of tin(IV) chloride for this cyclization was less efficient, producing alkene 28 only in 15% yield. Further manipulation of alkene 28 was used to complete the total synthesis of celastrol (29) in 31 steps overall.

The marine natural product (–)-cyclosmenospongine (32) is a tetracyclic meroterpenoid which was synthesized by Magauer and co-workers using polyene cyclizations (Scheme [7]).[12] The approach featured an unprecedented stepwise non-biomimetic polyene cyclization of an aryl enol ether 30 catalyzed by ethylaluminum dichloride as the Lewis acid. This led to the closure of three rings with excellent control of four stereocenters on a gram scale to yield alcohol 31 (83%) as a single diastereomer. Excellent stereoselectivity was observed due to the transition state governed by the double-bond geometry of the enol ether, thioenol ether, and epoxide. Utilizing the same strategy, (–)-5-epi-aureol (35) was also synthesized via alcohol 34 (91%), which was prepared from aryl enol ether 33.[10]

Chang and co-workers reported that tin(II) triflate catalyzed the polyene cyclization of β-keto sulfones (Scheme [8]).[13] It was found that this Lewis acid could catalyze the cyclization of β-keto sulfones 36 and 38, which were derived from geranyl bromide and farnesyl bromide, to give bi- and tricyclic sulfonyl dihydropyrans 37 and 39, respectively, as ambrox analogues via six-membered chairlike conformations.

Electrophilic polyene cyclization of zerumbone (40), an isoprenoid, via carbonyl activation was reported by Appendino and co-workers (Scheme [9]).[14] Carbonyl activation of zerumbone (40) with electrophilic reagents resulted in a Nazarov-type polyene cyclization, leading to the formation of a variety of cyclized products after intramolecular rearrangement. Zerumbone (40) is very stable towards mineral acids, but undergoes intramolecular cyclizations upon treatment with a variety of Lewis acids. Reaction of zerumbone (40) with aluminum trichloride resulted in the formation of the racemic tricyclic enone 41 (44%), featuring a 5/5/7 ring system as a single product along with recovered starting material. On the other hand, bicyclic dienone 42 (18%) was formed via tin(IV) chloride mediated cyclization. Furthermore, upon treatment of zerumbone (40) with boron tribromide, a mixture of isomeric hydrocarbons 43 (6%) and 44 (10%) were formed following a series of rearrangements reactions.


McErlean and co-workers reported the first total synthesis of 5-epi-taiwaniaquinone G (48) via Lewis acid catalyzed polyene cyclization (Scheme [10]).[15] Taiwaniaquinone G (49) is a diterpene containing a 6/5/6 trans-fused ring system. It was envisioned that Lewis acid mediated polyene cyclization of geranylbenzene 45 would lead to the formation of the desired trans-fused 5/6 ring system. Surprisingly, boron trifluoride–diethyl ether complex induced cationic cyclization led to the formation of a mixture of tricyclic products 46 and 47 in 50% combined yield, in which the cis-fused 5/6 ring system of tricyclic intermediate 46 was the major diastereomer (dr 7:1). The observed diastereoselectivity was further investigated by a computational study incorporating a conductor-like polarizable continuum model, which led to the conclusion that the observed selectivity was due to steric interactions that destabilized the transition state leading to the trans-configured diastereomer.
(–)-Parvifloron F (52) was found to be a valuable abietane diterpene that exhibits multiple biological activities such as antibacterial and antiproliferative activity against several human tumor cell lines.[16] Nakagawa-Goto and co-workers achieved the first total synthesis of (–)-parvifloron F (52), which utilized tin(IV) chloride mediated polyene cyclization of epoxide 50 to give alcohol 51 (60%), furnishing the tricyclic core of the natural product (Scheme [11]).

2.2
Brønsted Acid Mediated Polyene Cyclizations
Polyfunctionalized benzhydryl alcohols were found to be able to undergo SN1-type ionization thereby triggering polyene cyclization promoted by hot water and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) as a mildly acidic catalyst (Scheme [12]).[17] Stable carbocations, generated from such electron-rich benzylic alcohols by water, react with carbon, sulfur, and nitrogen nucleophiles in aqueous conditions. Qu and co-workers discovered that allylic alcohol 53 underwent polyene cyclization in a 3:1 mixture of refluxing water and HFIP to form octahydrophenanthrene product 54 (80%, dr 24:1:1:0.35) with excellent diastereoselectivity. Equivalent tricyclization of allylic alcohol 55 gave tetracycle 56 (51%, dr 3:1) in higher yield and with better diastereoselectivity when the reaction was performed in neat HFIP.
Further studies by the Qu group discovered that HFIP could act as a Brønsted acid (pK a = 9.3) catalyst to promote epoxide initiated cationic π-cyclizations (Scheme [13]).[18] The addition of tetraphenylphosphonium tetrafluoroborate enhanced the rate of reaction presumably due to trace amounts of hydrogen fluoride generated by tetrafluoroborate anion solvolysis in HFIP. Therefore, the reaction medium acted as both Brønsted acid and solvent to promote epoxides 57, 59, and 61 to undergo di-, tri-, and tetracyclizations rapidly to yield polycyclic compounds 58, 60, and 62, respectively.


Following the pioneering work on Lewis acid assisted chiral Brønsted acid (chiral LBA) mediated polyene cyclization from the Yamamoto and Corey groups,[19] Chein and co-workers reported an enantioselective polyene cyclization of 2-(homogeranyl)anisole 63 with the BINOL derivative 67 and tin(IV) chloride en route to galanal A (65) and B (66) (Scheme [14]).[20] Enantioselective protonation with the complex tin(IV) chloride·67 provided a mixture of tricyclic compound 64 along with monocyclized products as the major components. Subsequent exposure of the mixture to tin(IV) chloride and trifluoroacetic acid provided octahydrophenanthrene 64 (86% over 2 steps, 57% ee) as the key intermediate, which was converted into galanal A (65) and B (66).

Meroterpenoid berkeleyone A (70), which is produced by a fungus Penicillium rubrum, inhibits caspase-1, a protein associated with Alzheimer’s and Parkinson’s diseases. Newhouse and co-workers reported a 13-step total synthesis of (±)-berkeleyone A (70) utilizing a Brønsted acid mediated epoxy-diene polyene cyclization as the key step to form the tricyclic framework (Scheme [15]).[21] While most epoxide-initiated polyene cyclizations are activated with Lewis acids, the desired polyene cyclization of epoxide 68 required a Brønsted acid to facilitate the carbon-based nucleophilic attack of the β-keto ester moiety to terminate the cyclization. After extensive screening of Lewis and Brønsted acids promotors for the polyene cyclization of epoxide 68 to alcohol 69, it was found that the yellow ether solvated complex of HFeCl4, prepared in situ by reaction of iron(III) chloride in CH2Cl2 with anhydrous hydrogen chloride in diethyl ether, was the best promoter for the desired polyene cyclization to form alcohol 69 (39%). This led to the formation of three C–C bonds and generated six stereogenic centers to furnish the majority of the berkeleyone scaffold. Control experiments showed that the active Brønsted acid [FeCl4]– anion was superior to either iron(III) chloride or anhydrous hydrogen chloride alone.

Zhao and co-workers developed a chiral Brønsted acid N-phosphoramide from BINOL that catalyzed enantioselective polyene cyclization via protonation (Scheme [16]).[22] This methodology focused on the protonation of an imine to produce an iminium ion as initiator for the polyene cyclization. It was found that N-phosphoramidate 76 provided optimal enantioselectivity and addition of magnesium thiosulfate hexahydrate improved the reaction rate with enhanced enantioselectivity at –60 °C. A broad range of substituted arenes are compatible as terminating nucleophiles under the reaction conditions. Furthermore, its application was showcased in the first total synthesis of (–)-ferruginol (75), the enantiomer of diterpene natural product (+)-ferruginol which has antifungal, antimicrobial, antitumor, and anti-inflammatory activities. The N-phosphoramide 76 catalyzed enantioselective protonation of the resulting imine derived from aldehyde 73 with 4-toluenesulfonamide, which was utilized as the key step to induce polyene cyclization, forming the dicyclized intermediate 74 for the total synthesis of (–)-ferruginol (75).

A new protocol for the synthesis of cyclohexanones developed by Rodríguez and co-workers,[23] was extended to the cationic polyene cyclization of polyenynes (Scheme [17]). Tetrafluoroboric acid acts as the promotor for the cyclization, employing HFIP as the solvent. The reaction was initiated by alkene protonation and terminated by trapping the final alkenyl cation with water. Three different polyenynes derived from geraniol 77, nerol 79, and farnesol 81 were cyclized to provide ketones 78 (82%, dr >20:1), 80 (80%, dr 3:1), and 82 (65%, dr 3:1), respectively.

Qiu and co-workers have isolated new natural meroterpenoids (±)-cochlactone A (84) and B (85) that display anti-inflammatory activities (Scheme [18]).[24] It was postulated that the possible biogenetic pathway for cochlactone A (84) could be from ganomycin C (83) by acid-catalyzed cyclization. After optimization of the reaction conditions, it was found that exposure of ganomycin C (83) to p-toluenesulfonic acid resulted in polyene cyclization, thereby producing a product mixture containing (±)-cochlactone A (84) and B (85) (42% combined yield) in 2:1 ratio.

By utilizing sequential biomimetic polyketide and polyene cyclizations, the Barrett group completed a concise 6 step synthesis of (+)-hongoquercin A (89) (Scheme [19]).[7b] A farnesol (86)-derived resorcylate 87 was protonated enantioselectivity and regioselectively at the terminal alkene to promote polyene cyclization with tin(IV) chloride·90, developed by the Yamamoto group as a Lewis acid enhanced Brønsted acid,[19e] thereby producing meroterpenoid 88 (61%, 81% dr, 90% ee). Subsequent saponification of meroterpenoid 88 was used to complete the synthesis of (+)-hongoquercin A (89) (75%).

2.3
Halogen Electrophile Initiated Polyene Cyclizations

In 2009, the Snyder group invented bromodiethylsulfonium bromopentachloroantimonate (Et2SBr·SbCl5Br, BDSB, 93), which was shown to be an efficient reagent to induce bromonium ion mediated polyene cyclizations and electrophilic aromatic substitution reactions.[25] Recent findings by the Snyder group further demonstrated its ability to mediate the cyclization of alkene 90 to produce the spiro[5.5]undecane derivative 91 via bromonium-induced polyene cyclization accompanied by regio- and stereoselective α-bromination of the ketone functionality (Scheme [20]).[26] Spirocycle 91 possesses the essential framework of bromo-chamigrene 92, which could provide access to the chamigrene family of natural products.
The first reported catalytic enantioselective bromonium-mediated polyene cyclization was developed by the Yamamoto group (Scheme [21]).[27] This reaction was catalyzed by a chiral thiophosphoramidate 98 and employed 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 99) as an electrophilic bromine source. Enantioselective bromocyclization of geranylbenzenes 94 provided a mixture of fully and partially cyclized products. Subsequent treatment of the reaction mixture with chlorosulfonic acid promoted the final cyclization of the partially cyclized products to give bromides 95. On the other hand, bromocyclization of geranylphenols 96 under the same conditions provided fully cyclized bromides 97 as single diastereomers with excellent enantioselectivities without additional treatment with chlorosulfonic acid.

Rodríguez, Fañanás, and co-workers reported an acid-mediated cationic cyclization of polyenynes to form halogen-containing polycyclic compounds (Scheme [22]).[28] Protonation of the alkene functionality of enyne 77 by tetrafluoroboric acid induced a cationic polyene cyclization which was terminated by the alkyne group to form an intermediate alkenyl cation that reacted with a halide source. It was observed that the dienyne 77 cyclized to give the corresponding chloride 100 (76%), bromide 101 (70%), or fluoride 102 (90%) when the halogenated solvent used in the reaction was changed to dichloromethane, dibromomethane, or hexane. While a geraniol-derived dienyne gave the trans-fused compounds 100–102 exclusively, cis-fused compound 103 was obtained diastereoselectively from nerol-derived dienyne 79. Furthermore, cyclization with substrate 105 containing an internal alkyne produced an indane derivative 106 (70%) as a mixture of geometric isomers. It is important to note that bicyclic bromide 101 served as a common intermediate for the total synthesis of (±)-austrodoral (107), (±)-pallescensin A (108), and (±)-9-epi-ambrox (109).


Hennecke and co-workers discovered that a stable bromonium salt 111 underwent electrophilic addition to alkenes that induced halocyclization of polyenes (Scheme [23]).[29] Bromonium salt 111 is a sterically demanding bromonium ion with the weakly coordinating tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (BArF–) counterion. Reaction of this salt 111 with homogeranyl arenes 110 effectively furnished fully cyclized bromides 112–114 (29–64%).
The halocyclization of polyenes was also mediated using an electrophilic halogen source in the presence of the Lewis base morpholine with HFIP acting as the Lewis acid according to findings in 2018 by Gulder and co-workers (Scheme [24]).[30] Reaction of morpholine and either N-bromosuccinimide (NBS), N-iodosuccinimide (NIS), or 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) generates the corresponding N-halomorpholine 116, which acts as the halogenating reagent. Homogeranyl benzene 115 was successfully converted into the corresponding fully cyclized halides 117–119 with excellent diastereoselectivities using N-halomorpholines 116. HFIP was identified as the best solvent for this transformation because of its Lewis acidic, highly polar, and strongly hydrogen-bond donating, but weakly nucleophilic, properties. It is essential for HFIP to activate the N-halomorpholine 116 via hydrogen-bonding for halogenation to take place. Furthermore, HFIP could possibly prearrange the aryl-diene 115 via hydrophobic interactions to facilitate the cyclization. Lastly, the Lewis acidity of HFIP allows activation of the halonium ion intermediate to induce polyene cyclization.

2.4
Sulfur Electrophile Initiated Polyene Cyclizations
The Snyder group reported three electrophilic alkyldisulfanium salts that are capable of inducing sulfur-mediated cationic polyene cyclizations (Scheme [25]).[31] Alkyldisulfanium salts 120–122, effectively serving as electrophilic sources of ethylsulfanium, methylsulfanium, and 3,3,3-trifluoropropylsulfanium cations, respectively, mediate the cyclization of polyene 123 to produce octahydrophenanthrenes 124–126 in modest yields (16–52%).

Further investigations led to the development of alkyl- and aryldisulfanium salts 127 that could induce thiiranium-mediated polyene cyclizations (Scheme [26]).[32] Cyclization of homogeranylbenzene 115 with alkyl and aryl sulfide electrophiles produced sulfides 128 with improved yields compared to those obtained with alkyldisulfanium salts 120–122.

The first catalytic and enantioselective thiiranium ion mediated polyene cyclization was reported by Denmark and co-workers (Scheme [27]).[33] Homogeranylarenes 94 and ortho-geranylphenols 96 were cyclized to give the corresponding polycyclic compounds 129 and 130, respectively, via the intermediacy of an enantiomerically enriched thiiranium ion in HFIP. This reaction was catalyzed by a chiral Lewis base 136 and sulfenylating reagent 135. It is important to note that the use of HFIP as the solvent is crucial to suppress sulfenylation of the internal alkene due to solvophobic interactions between the highly polar HFIP medium and the lipophilic polyene substrate, minimizing exposure of internal alkenes. Furthermore, the acidity of HFIP is beneficial to generate the cationic active catalyst. Application of the methodology was used in enantioselective total syntheses of (+)-ferruginol (133) and (+)-hinokiol (134) from sulfide 132.
2.5
Transition-Metal-Mediated Cationic Polyene Cyclizations
Following the pioneering work by Gagné and co-workers on electrophilic platinum-complex-catalyzed polyene cyclization reactions,[4c] in 2014 they developed a new platinum(II) complex 143 with an N-heterocyclic carbene (NHC) containing pincer ligand for the synthesis of sterol-like polycyclic compounds via sequential electrophilic polyene cyclization and oxidative protonolysis (Scheme [28]).[34] The terminal alkene of the substrates is activated by the electrophilic platinum(II) complex to initiate the polyene cyclization, followed by protodemetalation to regenerate the Pt catalyst. This methodology has a broad substrate scope and is able to induce di-, tri-, and tetracyclization reactions with a range of polyene substrates 137, 139, and 141. Diphenylamine was added as a proton shuttle to enhance the reaction rate in some cases.



The platinum(II) catalyst 143 was also used in a study on the influence of alkene substitution on pentaene cyclizations (Scheme [29]).[35] Three unique analogues (144, 146, and 148) of pentaenes 141, with different methyl group modification, were synthesized and treated with catalyst 143, inducing polyene cyclizations to provide the cyclized products 145, 147, and 150/151. It was found that replacement of methyl groups by hydrogen did not affect the efficiency of polyene cyclization compared to that of 141 except to diminish reaction rates. Pentaenes 144 and 146 both underwent the anticipated cyclization to give alkene 145 (18%) and 147 (54%), respectively. On the other hand, cyclization of pentaene 148 provided a mixture of two tetracyclic compounds 150 (32%) and 151 (20%), which were formed from the common intermediate 149 via hydride shifts. Kinetic studies showed that the rate of pentaene cyclization of substrates 141, 144, 146, and 148 follows the order 141 > 148 > 146 > 144. Computational studies suggest that pentaenes 141 and 144 form the first three rings in a concerted mechanism to generate a carbocation intermediate for further cyclizations. Pentaene 146 forms the cyclohexyl ring first, followed by concerted cyclizations to close the second and third rings. Pentaene 148 first undergoes concerted cyclizations to form the first two rings, followed by stepwise cyclizations to yield products 150 and 151 after different hydride shifts. Pentaenes 141, 144, and 146 generate tertiary carbocations while pentaene 148 generates a secondary carbocation after the concerted cyclizations. Furthermore, intrinsic reaction coordinate (IRC) calculations for the cyclization of pentaene 144 indicated that the reaction coordinates of the platinum(II)-catalyzed pentaene cyclizations are similar to those of the enzymatic cyclizations of squalene and oxidosqualene, suggesting that it is essentially an organometallic equivalent of a key biomimetic process.

Building upon previous research on gold(I)-catalyzed polyene cyclizations,[36] a new advance was reported by Echavarren and Rong (Scheme [30]).[37] Activation of the enyne of polyene substrates by gold(I) triggered polyene cyclization to produce steroid-like compounds. 1,5-Enyne 152 cyclized to give alkene 154 (79%) with the arene acting as the terminating nucleophile. Additional examples using alcohols, phenols, and heteroarenes as the terminal nucleophiles were also successfully cyclized. Furthermore, di- and tricyclization of polyenynes 156 and 160 gave the tri- and tetracyclic compounds 158 (90%) and 162 (59%), respectively, under the same conditions. It was also found that bromoalkynes could be used as initiators of polyene cyclization to give cyclic bromoalkenes. For instance, the bromoalkyne analogues 153, 157, and 161 were cyclized to give the corresponding synthetically useful bromoalkenes 155 (92%), 159 (84%), and 163 (51%). The use of chiral gold(I) catalysts for the enantioselective variant of this cyclization was also investigated. It was found that moderate enantioselectivity was observed when the dinuclear gold(I) complex of BIPHEP 165 and silver(I) ditriflylimide were employed in the cyclization of 152 into 154 (85%, 54% ee).

(–)-Insulicolide A (168) is a nitrobenzoyloxy-substituted sesquiterpenoids with promising anticancer activity against several human cancer cell lines. The first total synthesis of (–)-insulicolide A (168) and related sesquiterpenoids was accomplished by Yang, Chen, and co-workers (Scheme [31]).[38] A key intermediate 167 in this synthesis was prepared from allylic alcohol 166 by an iridium-catalyzed enantioselective polyene cyclization reaction, developed by Carreira and co-workers,[39] in the presence of ligand 169 to furnish the drimane core 167 (73%, >99% ee).

The same strategy was utilized by Li and co-workers in the total synthesis of (–)-septedine (172) and (–)-7-deoxyseptedine (173) (Scheme [32]).[40] The key enantiomerically pure alkene 171 (61%, >99% ee) was prepared by the iridium-catalyzed polyene cyclization of allylic alcohol 170 employing phosphoramidite 174 as the chiral ligand.
3
Radical Polyene Cyclizations
3.1Transition-Metal-Mediated Radical Polyene Cyclizations
While classical cationic polyene cyclizations dominate the literature on this topic, radical-mediated polyene cyclizations were proposed as early as the 1960s by Breslow.[41] The hypothesis of the biosynthetic pathway for polyene cyclizations was ambiguous, which led Breslow to the formulation of a radical approach, which was subsequently rejected. In contrast to cationic polyene cyclizations, radical processes allow a milder approach towards functionalized decalins, as radicals operate on a lower relative energy level than cationic intermediates. It should be noted that radical mediators can transform highly functionalized polyene precursors into trans-decalins with remarkable stereocontrol in a non-concerted mechanism that could be rationalized in terms of the Beckwith–Houk rules.[42]
In 2015, Oltra, Rosales, and co-workers reported the synthesis of (±)-aureol (178), a marine natural product which exhibits cytotoxicity against A549 human non-small cell lung cancer cells and anti-influenza-A virus activity (Scheme [33]).[43] Application of a titanocene(III)-mediated reductive polyene cyclization of racemic epoxide 176, derived from alcohol 175, provided alcohol 177 with an exocyclic alkene with excellent relative stereochemical control. Subsequent boron trifluoride–diethyl ether catalyzed rearrangement was used in the transformation of alcohol 177 into (±)-aureol (178).

The synthetic power of the titanocene(III)-mediated polyene cyclization was further demonstrated by Takahashi and co-workers in the synthesis of (–)-ent-pyripyropene A (181) (Scheme [34]).[44] Titanocene(III)-induced polyene cyclization of epoxide 179 gave alcohol 180 (60%). The use of single enantiomer epoxide 179 thereby controls the outcome of the formation of 4 contiguous stereocenters. The choice of ligand in the asymmetric epoxidation allows diversification towards natural and unnatural derivatives of the pyripyropene family. The titanocene(III)-mediated cyclization was fully automated, reproducibly allowing access to 180, a common intermediate in terpene natural product synthesis.

Seifert and Göhl utilized a similar strategy in the synthesis of (–)-3-oxotauranin (184) and (–)-3β-hydroxytauranin (185) (Scheme [35]).[45] Titanocene(III)-mediated radical polyene cyclization of epoxide 182 afforded alcohol 183, subsequent global deprotection and adjustment of oxidation state completed the synthesis of (–)-3-oxotauranin (184) and (–)-3β-hydroxytauranin (185).

In 2015, Trotta reported the cyclization of a geranyl β-keto ester 186 under radical conditions employing a Mn(III)/Cu(II) system to provide alcohol 187 (50%) (Scheme [36]).[46] This allowed the preparation of the terpenoid trans-decalin core of (±)-oridamycin A (188) and B (189) with the methyl ester moiety in the axial position.

Li and co-workers reported the use of a titanocene(III)-mediated polyene cyclization of epoxide 190, furnishing the trans-decalin core 191 (34%), with the alcohol in equatorial and the methyl ester in equatorial positions (Scheme [37]).[47] Subsequent transformation of alcohol 191 completed the divergent synthesis of (–)-sespenine (192) and (+)-xiamycin A (193).

In 2017, Shoji and co-workers reported a radical cyclization of β-keto ester 194 which was mediated by Mn(III)/Cu(II) to give tetracyclic ketone 196 (11%) and tricyclic ketone 197, as a mixture of regioisomers (22%) via intermediate 195 (Scheme [38]).[48] The functionalization arose from tuning the electron-donating or -demanding properties of the polyene scaffold to allow efficient cyclization between alternating electrophilic radicals with electron-rich trisubstituted olefins.

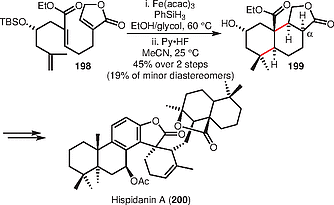
In 2017, Liu and co-workers reported the synthesis of (–)-hispidanin A (200), a natural product with promising anticancer properties, via a hydrogen atom transfer (HAT) mediated triene cyclization to construct the tricyclic core of (–)-hispidanin A (200) (Scheme [39]).[49] The use of iron(III) acetylacetonate and triphenylsilane as hydrogen source smoothly furnished tricyclic lactone 199 from alkene 198 (45% over 2 steps) after deprotection, thereby establishing two quaternary and four contiguous stereocenters in one step. In a 2018 follow-up paper, Liu and co-workers reported further mechanistic studies, producing deuteration at the α-position of the lactone, supporting the hypothesis that the reaction involves the formation of an enolate anion.[50]
While most of these transformations are initiated by a radical generated from metal complexes, in 2018 the Baran group exploited electrochemical initiation for a radical polyene cyclization in the divergent synthesis of pyrone diterpenes sesquicillin A (205), higginsianin A (206), and subglutinol A (203) and B (204) (Scheme [40]).[51] Cyclization of β-keto ester 201 provided key intermediate alkene 202 (42%) which was further derivatized into the different diterpene natural products.

3.2
Photocatalyst-Mediated Polyene Cyclizations

Photoredox catalysis has been an ever-growing field over the last decade with numerous examples of synthetic transformations, and applications in natural product or pharmaceutical synthesis. Since the seminal work published by Demuth and co-workers on photochemically induced electron transfer polyene cyclizations,[52] the utility of photocatalyst-mediated polyene cyclization has remained underexplored. One noticeable example of polyene cyclization catalyzed with eosin Y (213) was published in 2015 by Zhang, Luo, and co-workers (Scheme [41]).[53] The mild photoredox-catalyzed transformation tolerated various functionalization and the cyclization proceeded with excellent relative stereochemical control and diastereoselectivity for various ring systems. A selection of solvents and organic and inorganic photoredox catalysts were examined and optimal results were obtained with eosin Y in HFIP. Subsequently, an array of geranylphenols 207 and farnesylphenols 209 with various functional groups in the para position were cyclized to produce chromanes 208 and 210, respectively. 1,3-Diketones 211 also underwent photocatalyzed polyene cyclizations, thereby producing chromenes 212 with various substituents R1 and R2 of different electronic nature.
The synthetic utility of the photocatalyst-initiated polyene cyclization was further demonstrated in the 7-step synthesis of (±)-hongoquercin A (89) (Scheme [42]).[54] Cyclization of phenol 214 catalyzed by eosin Y (213) provided tetracyclic compound 215 (60% by NMR), a key intermediate with the complete carbon framework of the natural product.


Trotta reported attempts to employ photoredox catalysts as polyene cyclization initiators during his studies on the total synthesis of xiamycin A (193).[55] While earlier work on (±)-oridamycin A (188) showed that manganese(III)-mediated cyclization gave the trans-decalin 187 with the methoxy ester in the axial position (Scheme [43]); photoredox catalysts were able to convert an α-bromo-β-keto ester 216 into the same trans-decalin compound 187. When the acylated linear precursor was subjected to these conditions, it resulted in complete decomposition. Although the yields were low, the reaction proceeded at catalyst loadings as low as 0.01 mol%.
4
Origin of Stereocontrol in Polyene Cyclizations
Several general remarks on the observed stereoselectivity of the various methods of cationic polyene cyclization reactions can be made. The underlying principle of all the cationic cyclization reactions mentioned is that the prearranged pseudo-chair-chair-(chair) conformation such as of polyene 217 or 218 allows the efficient preparation of polycyclic compound 221 with predefined relative stereochemistry. This is based on the stereoelectronic arrangement of an olefinic double bond in close proximity to the electrophilic carbenium center in intermediate 219 and subsequently nucleophilic attack by an olefinic bond further down the chain on the intermediately formed carbenium center (Scheme [44]). As such the trans-substitution of the polyene precursor sets the outcome for the observed trans-decalin stereoselectivity in the concerted cationic cyclization through the given prearranged conformation according to the Stork–Eschenmoser hypothesis.[1a] [b] In cases where the reactions are not proceeding in a concerted fashion, a mixture of products are formed.[1a]


On the other hand, the origin of stereocontrol in radical polyene cyclization reactions could be rationalized by the Beckwith–Houk rules in addition to the Stork–Eschenmoser hypothesis (Scheme [45]).[42] As an illustration, titanium(III)-mediated cyclization of epoxide 222 would presumably proceed in a non-concerted fashion via radical intermediates 223 and 224 and favored 6-endo-trig cyclization via a chair transition state over 5-exo-trig cyclization to produce alcohol 225 due to ring strain.
5
Conclusion
Inspired by nature’s efficient and highly stereochemically controlled syntheses of primary and secondary metabolites via polyene cyclizations, the use of initiators such as Lewis acids, metal complexes, halonium ion transfer reagents, sulfanium ion transfer reagents, and Brønsted acids have demonstrated the synthetic utility of these bioinspired transformations with comparable selectivities and efficiencies to the enzymatic processes. Recent developments of novel polyene cyclization methodologies have significantly expanded the scope of terpene and polyene functionalization reactions, providing a wide range of unique carbon frameworks. Radical-mediated polyene cyclizations have been further investigated to achieve different modes of cyclization from the cationic pathway, allowing early and late stage functionalization of polyenes for terpene and meroterpenoid total syntheses. Photocatalyst-mediated polyene cyclizations have emerged as new alternatives, which employ milder reaction conditions as well as lower catalyst loadings. It goes without saying that future developments in electrophilic reagents and catalysts will further advance the synthetic utility of polyene cyclizations with excellent enantio- and diastereoselectivities, greatly impacting the area of terpenoid, meroterpenoid, and steroid total syntheses amongst other classes of compounds.
Acknowledgment
We thank GlaxoSmithKline for the endowment (to A.G.M.B) as well as Drs. Alfred and Isabel Bader for their additional support. We thank Dr. Sara R. Goldstein and Dr. Joshua Almond-Thynne for proofreading this review article.
-
References
- 1a Stork G, Burgstahler AW. J. Am. Chem. Soc. 1955; 77: 5068
- 1b Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv. Chim. Acta 1955; 38: 1890
- 1c Eschenmoser A, Arigoni D. Helv. Chim. Acta 2005; 88: 3011
- 2a Johnson WS, Semmelhack MF, Sultanbawa MU. S, Dolak LA. J. Am. Chem. Soc. 1968; 90: 2994
- 2b Johnson WS. Acc. Chem. Res. 1968; 1: 1
- 2c Johnson WS. Bioorg. Chem. 1976; 5: 51
- 4a Snyder SA, Levinson AM. Comprehensive Organic Synthesis II . Vol. 3. Elsevier; Oxford: 2014: 268
- 4b Li X.-W, Nay B. Nat. Prod. Rep. 2014; 31: 533
- 4c Felix RJ, Munro-Leighton C, Gagné MR. Acc. Chem. Res. 2014; 47: 2319
- 4d Yoshimura T. Tetrahedron Lett. 2014; 55: 5109
- 4e Dhambri S, Mohammad S, Van Buu ON, Galvani G, Meyer Y, Lannou M.-I, Sorin G, Ardisson J. Nat. Prod. Rep. 2015; 32: 841
- 4f Ardkhean R, Caputo DF. J, Morrow SM, Shi H, Xiong Y, Anderson EA. Chem. Soc. Rev. 2016; 45: 1557
- 4g Ungarean CN, Southgate EH, Sarlah D. Org. Biomol. Chem. 2016; 14: 5454
- 4h Hung K, Hu X, Maimone TJ. Nat. Prod. Rep. 2018; 35: 174
- 4i Landry ML, Burns NZ. Acc. Chem. Res. 2018; 51: 1260
- 5a Xu H, Tang H, Feng H, Li Y. Tetrahedron Lett. 2014; 55: 7118
- 5b Xu H, Tang H, Feng H, Li Y. J. Org. Chem. 2014; 79: 10110
- 6 Surendra K, Qiu W, Corey EJ. J. Am. Chem. Soc. 2011; 133: 9724
- 7a Barrett TN, Barrett AG. M. J. Am. Chem. Soc. 2014; 136: 17013
- 7b Ma T.-K, Elliott DC, Reid S, White AJ. P, Parsons PJ, Barrett AG. M. J. Org. Chem. 2018; 83: 13276
- 8 Rosen BR, Werner EW, O’Brien AG, Baran PS. J. Am. Chem. Soc. 2014; 136: 5571
- 9 Suzuki K, Yamakoshi H, Nakamura S. Chem. Eur. J. 2015; 21: 17605
- 10 Rajendar G, Corey EJ. J. Am. Chem. Soc. 2015; 137: 5837
- 11 Camelio AM, Johnson TC, Siegel D. J. Am. Chem. Soc. 2015; 137: 11864
- 12a Speck K, Wildermuth R, Magauer T. Angew. Chem. Int. Ed. 2016; 55: 14131
- 12b Speck K, Magauer T. Chem. Eur. J. 2017; 23: 1157
- 13 Chan C.-K, Chen Y.-H, Chang M.-Y. Tetrahedron 2016; 72: 5121
- 14 Minassi A, Pollastro F, Chianese G, Caprioglio D, Taglialatela-Scafati O, Appendino G. Angew. Chem. Int. Ed. 2017; 56: 7935
- 15 Graham M, Baker RW, McErlean CS. P. Eur. J. Org. Chem. 2017; 908
- 16 Saito Y, Goto M, Nakagawa-Goto K. Org. Lett. 2018; 20: 628
- 17 Zhang F.-Z, Tian Y, Li G.-X, Qu J. J. Org. Chem. 2015; 80: 1107
- 18 Tian Y, Xu X, Zhang L, Qu J. Org. Lett. 2016; 18: 268
- 19a Ishihara K, Nakamura S, Yamamoto H. J. Am. Chem. Soc. 1999; 121: 4906
- 19b Nakamura S, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2000; 122: 8131
- 19c Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2001; 123: 1505
- 19d Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2002; 124: 3647
- 19e Ishibashi H, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2004; 126: 11122
- 19f Surendra K, Corey EJ. J. Am. Chem. Soc. 2012; 134: 11992
- 19g Surendra K, Rajendar G, Corey EJ. J. Am. Chem. Soc. 2014; 136: 642
- 20 Lin S.-C, Chein R.-J. J. Org. Chem. 2017; 82: 1575
- 21 Elkin M, Szewczyk SM, Scruse AC, Newhouse TR. J. Am. Chem. Soc. 2017; 139: 1790
- 22 Fan L, Han C, Li X, Yao J, Wang Z, Yao C, Chen W, Wang T, Zhao J. Angew. Chem. Int. Ed. 2018; 57: 2115
- 23 Alonso P, Fontaneda R, Pardo P, Fañanás FJ, Rodríguez F. Org. Lett. 2018; 20: 1659
- 24 Peng X.-R, Lu S.-Y, Shao L.-D, Zhou L, Qiu M.-H. J. Org. Chem. 2018; 83: 5516
- 25a Snyder SA, Treitler DS. Angew. Chem. Int. Ed. 2009; 48: 7899
- 25b Snyder SA, Treitler DS, Brucks AP. J. Am. Chem. Soc. 2010; 132: 14303
- 26 Shen M, Kretschmer M, Brill ZG, Snyder SA. Org. Lett. 2016; 18: 5018
- 27 Samanta RC, Yamamoto H. J. Am. Chem. Soc. 2017; 139: 1460
- 28 Alonso P, Pardo P, Fontaneda R, Fañanás FJ, Rodríguez F. Chem. Eur. J. 2017; 23: 13158
- 29 Ascheberg C, Bock J, Buß F, Mück-Lichtenfeld C, Daniliuc CG, Bergander K, Dielmann F, Hennecke U. Chem. Eur. J. 2017; 23: 11578
- 30 Arnold AM, Pöthig A, Drees M, Gulder T. J. Am. Chem. Soc. 2018; 140: 4344
- 31 Schevenels FT, Shen M, Snyder SA. Org. Lett. 2017; 19: 2
- 32 Cole C, Chi H, DeBacker K, Snyder S. Synthesis 2018; 50: A-H
- 33 Tao Z, Robb KA, Zhao K, Denmark SE. J. Am. Chem. Soc. 2018; 140: 3569
- 34 Geier MJ, Gagné MR. J. Am. Chem. Soc. 2014; 136: 3032
- 35 McCulley CH, Geier MJ, Hudson BM, Gagné MR, Tantillo DJ. J. Am. Chem. Soc. 2017; 139: 11158
- 36a Toullec PY, Blarre T, Michelet V. Org. Lett. 2009; 11: 2888
- 36b Sethofer SG, Mayer T, Toste FD. J. Am. Chem. Soc. 2010; 132: 8276
- 36c Pradal A, Chen Q, Faudot dit Bel P, Toullec P, Michelet V. Synlett 2012; 2012: 74
- 37 Rong Z, Echavarren AM. Org. Biomol. Chem. 2017; 15: 2163
- 38 Lai Y, Zhang N, Zhang Y, Chen J.-H, Yang Z. Org. Lett. 2018; 20: 4298
- 39 Schafroth MA, Sarlah D, Krautwald S, Carreira EM. J. Am. Chem. Soc. 2012; 134: 20276
- 40 Zhou S, Guo R, Yang P, Li A. J. Am. Chem. Soc. 2018; 140: 9025
- 41a Breslow R, Barrett E, Mohacsi E. Tetrahedron Lett. 1962; 3: 1207
- 41b Breslow R, Olin SS, Groves JT. Tetrahedron Lett. 1968; 9: 1837
- 42 Curran DP, Porter NA, Giese B. Stereochemistry of Radical Reactions . VCH; Weinheim: 1995
- 43 Rosales A, Muñoz-Bascón J, Roldan-Molina E, Rivas-Bascón N, Padial NM, Rodríguez-Maecker R, Rodríguez-García I, Oltra JE. J. Org. Chem. 2015; 80: 1866
- 44 Fuse S, Ikebe A, Oosumi K, Karasawa T, Matsumura K, Izumikawa M, Johmoto K, Uekusa H, Shin-ya K, Doi T, Takahashi T. Chem.–Eur. J. 2015; 21: 9454
- 45 Göhl M, Seifert K. Eur. J. Org. Chem. 2015; 6249
- 46 Trotta AH. Org. Lett. 2015; 17: 3358
- 47 Sun Y, Meng Z, Chen P, Zhang D, Baunach M, Hertweck C, Li A. Org. Chem. Front. 2016; 3: 368
- 48 Furuta M, Hanaya K, Sugai T, Shoji M. Tetrahedron 2017; 73: 2316
- 49 Deng H, Cao W, Liu R, Zhang Y, Liu B. Angew. Chem. Int. Ed. 2017; 56: 5849
- 50 Cao W, Deng H, Sun Y, Liu B, Qin S. Chem. Eur. J. 2018; 24: 9120
- 51 Merchant RR, Oberg KM, Lin Y, Novak AJ. E, Felding J, Baran PS. J. Am. Chem. Soc. 2018; 140: 7462
- 52a Hoffmann U, Gao Y, Pandey B, Klinge S, Warzecha K.-D, Krueger C, Roth HD, Demuth M. J. Am. Chem. Soc. 1993; 115: 10358
- 52b Heinemann C, Demuth M. J. Am. Chem. Soc. 1997; 119: 1129
- 52c Heinemann C, Demuth M. J. Am. Chem. Soc. 1999; 121: 4894
- 52d Goeller F, Heinemann C, Demuth M. Synthesis 2001; 1114
- 52e Rosales V, Zambrano J, Demuth M. Eur. J. Org. Chem. 2004; 1798
- 52f Ozser ME, Icil H, Makhynya Y, Demuth M. Eur. J. Org. Chem. 2004; 3686
- 53 Yang Z, Li H, Zhang L, Zhang M.-T, Cheng J.-P, Luo S. Chem. Eur. J. 2015; 21: 14723
- 54 Yang Z, Li S, Luo S. Acta Chim. Sin. 2017; 75: 351
- 55 Trotta AH. J. Org. Chem. 2017; 82: 13500
For recent reviews in this area, see
-
References
- 1a Stork G, Burgstahler AW. J. Am. Chem. Soc. 1955; 77: 5068
- 1b Eschenmoser A, Ruzicka L, Jeger O, Arigoni D. Helv. Chim. Acta 1955; 38: 1890
- 1c Eschenmoser A, Arigoni D. Helv. Chim. Acta 2005; 88: 3011
- 2a Johnson WS, Semmelhack MF, Sultanbawa MU. S, Dolak LA. J. Am. Chem. Soc. 1968; 90: 2994
- 2b Johnson WS. Acc. Chem. Res. 1968; 1: 1
- 2c Johnson WS. Bioorg. Chem. 1976; 5: 51
- 4a Snyder SA, Levinson AM. Comprehensive Organic Synthesis II . Vol. 3. Elsevier; Oxford: 2014: 268
- 4b Li X.-W, Nay B. Nat. Prod. Rep. 2014; 31: 533
- 4c Felix RJ, Munro-Leighton C, Gagné MR. Acc. Chem. Res. 2014; 47: 2319
- 4d Yoshimura T. Tetrahedron Lett. 2014; 55: 5109
- 4e Dhambri S, Mohammad S, Van Buu ON, Galvani G, Meyer Y, Lannou M.-I, Sorin G, Ardisson J. Nat. Prod. Rep. 2015; 32: 841
- 4f Ardkhean R, Caputo DF. J, Morrow SM, Shi H, Xiong Y, Anderson EA. Chem. Soc. Rev. 2016; 45: 1557
- 4g Ungarean CN, Southgate EH, Sarlah D. Org. Biomol. Chem. 2016; 14: 5454
- 4h Hung K, Hu X, Maimone TJ. Nat. Prod. Rep. 2018; 35: 174
- 4i Landry ML, Burns NZ. Acc. Chem. Res. 2018; 51: 1260
- 5a Xu H, Tang H, Feng H, Li Y. Tetrahedron Lett. 2014; 55: 7118
- 5b Xu H, Tang H, Feng H, Li Y. J. Org. Chem. 2014; 79: 10110
- 6 Surendra K, Qiu W, Corey EJ. J. Am. Chem. Soc. 2011; 133: 9724
- 7a Barrett TN, Barrett AG. M. J. Am. Chem. Soc. 2014; 136: 17013
- 7b Ma T.-K, Elliott DC, Reid S, White AJ. P, Parsons PJ, Barrett AG. M. J. Org. Chem. 2018; 83: 13276
- 8 Rosen BR, Werner EW, O’Brien AG, Baran PS. J. Am. Chem. Soc. 2014; 136: 5571
- 9 Suzuki K, Yamakoshi H, Nakamura S. Chem. Eur. J. 2015; 21: 17605
- 10 Rajendar G, Corey EJ. J. Am. Chem. Soc. 2015; 137: 5837
- 11 Camelio AM, Johnson TC, Siegel D. J. Am. Chem. Soc. 2015; 137: 11864
- 12a Speck K, Wildermuth R, Magauer T. Angew. Chem. Int. Ed. 2016; 55: 14131
- 12b Speck K, Magauer T. Chem. Eur. J. 2017; 23: 1157
- 13 Chan C.-K, Chen Y.-H, Chang M.-Y. Tetrahedron 2016; 72: 5121
- 14 Minassi A, Pollastro F, Chianese G, Caprioglio D, Taglialatela-Scafati O, Appendino G. Angew. Chem. Int. Ed. 2017; 56: 7935
- 15 Graham M, Baker RW, McErlean CS. P. Eur. J. Org. Chem. 2017; 908
- 16 Saito Y, Goto M, Nakagawa-Goto K. Org. Lett. 2018; 20: 628
- 17 Zhang F.-Z, Tian Y, Li G.-X, Qu J. J. Org. Chem. 2015; 80: 1107
- 18 Tian Y, Xu X, Zhang L, Qu J. Org. Lett. 2016; 18: 268
- 19a Ishihara K, Nakamura S, Yamamoto H. J. Am. Chem. Soc. 1999; 121: 4906
- 19b Nakamura S, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2000; 122: 8131
- 19c Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2001; 123: 1505
- 19d Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2002; 124: 3647
- 19e Ishibashi H, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2004; 126: 11122
- 19f Surendra K, Corey EJ. J. Am. Chem. Soc. 2012; 134: 11992
- 19g Surendra K, Rajendar G, Corey EJ. J. Am. Chem. Soc. 2014; 136: 642
- 20 Lin S.-C, Chein R.-J. J. Org. Chem. 2017; 82: 1575
- 21 Elkin M, Szewczyk SM, Scruse AC, Newhouse TR. J. Am. Chem. Soc. 2017; 139: 1790
- 22 Fan L, Han C, Li X, Yao J, Wang Z, Yao C, Chen W, Wang T, Zhao J. Angew. Chem. Int. Ed. 2018; 57: 2115
- 23 Alonso P, Fontaneda R, Pardo P, Fañanás FJ, Rodríguez F. Org. Lett. 2018; 20: 1659
- 24 Peng X.-R, Lu S.-Y, Shao L.-D, Zhou L, Qiu M.-H. J. Org. Chem. 2018; 83: 5516
- 25a Snyder SA, Treitler DS. Angew. Chem. Int. Ed. 2009; 48: 7899
- 25b Snyder SA, Treitler DS, Brucks AP. J. Am. Chem. Soc. 2010; 132: 14303
- 26 Shen M, Kretschmer M, Brill ZG, Snyder SA. Org. Lett. 2016; 18: 5018
- 27 Samanta RC, Yamamoto H. J. Am. Chem. Soc. 2017; 139: 1460
- 28 Alonso P, Pardo P, Fontaneda R, Fañanás FJ, Rodríguez F. Chem. Eur. J. 2017; 23: 13158
- 29 Ascheberg C, Bock J, Buß F, Mück-Lichtenfeld C, Daniliuc CG, Bergander K, Dielmann F, Hennecke U. Chem. Eur. J. 2017; 23: 11578
- 30 Arnold AM, Pöthig A, Drees M, Gulder T. J. Am. Chem. Soc. 2018; 140: 4344
- 31 Schevenels FT, Shen M, Snyder SA. Org. Lett. 2017; 19: 2
- 32 Cole C, Chi H, DeBacker K, Snyder S. Synthesis 2018; 50: A-H
- 33 Tao Z, Robb KA, Zhao K, Denmark SE. J. Am. Chem. Soc. 2018; 140: 3569
- 34 Geier MJ, Gagné MR. J. Am. Chem. Soc. 2014; 136: 3032
- 35 McCulley CH, Geier MJ, Hudson BM, Gagné MR, Tantillo DJ. J. Am. Chem. Soc. 2017; 139: 11158
- 36a Toullec PY, Blarre T, Michelet V. Org. Lett. 2009; 11: 2888
- 36b Sethofer SG, Mayer T, Toste FD. J. Am. Chem. Soc. 2010; 132: 8276
- 36c Pradal A, Chen Q, Faudot dit Bel P, Toullec P, Michelet V. Synlett 2012; 2012: 74
- 37 Rong Z, Echavarren AM. Org. Biomol. Chem. 2017; 15: 2163
- 38 Lai Y, Zhang N, Zhang Y, Chen J.-H, Yang Z. Org. Lett. 2018; 20: 4298
- 39 Schafroth MA, Sarlah D, Krautwald S, Carreira EM. J. Am. Chem. Soc. 2012; 134: 20276
- 40 Zhou S, Guo R, Yang P, Li A. J. Am. Chem. Soc. 2018; 140: 9025
- 41a Breslow R, Barrett E, Mohacsi E. Tetrahedron Lett. 1962; 3: 1207
- 41b Breslow R, Olin SS, Groves JT. Tetrahedron Lett. 1968; 9: 1837
- 42 Curran DP, Porter NA, Giese B. Stereochemistry of Radical Reactions . VCH; Weinheim: 1995
- 43 Rosales A, Muñoz-Bascón J, Roldan-Molina E, Rivas-Bascón N, Padial NM, Rodríguez-Maecker R, Rodríguez-García I, Oltra JE. J. Org. Chem. 2015; 80: 1866
- 44 Fuse S, Ikebe A, Oosumi K, Karasawa T, Matsumura K, Izumikawa M, Johmoto K, Uekusa H, Shin-ya K, Doi T, Takahashi T. Chem.–Eur. J. 2015; 21: 9454
- 45 Göhl M, Seifert K. Eur. J. Org. Chem. 2015; 6249
- 46 Trotta AH. Org. Lett. 2015; 17: 3358
- 47 Sun Y, Meng Z, Chen P, Zhang D, Baunach M, Hertweck C, Li A. Org. Chem. Front. 2016; 3: 368
- 48 Furuta M, Hanaya K, Sugai T, Shoji M. Tetrahedron 2017; 73: 2316
- 49 Deng H, Cao W, Liu R, Zhang Y, Liu B. Angew. Chem. Int. Ed. 2017; 56: 5849
- 50 Cao W, Deng H, Sun Y, Liu B, Qin S. Chem. Eur. J. 2018; 24: 9120
- 51 Merchant RR, Oberg KM, Lin Y, Novak AJ. E, Felding J, Baran PS. J. Am. Chem. Soc. 2018; 140: 7462
- 52a Hoffmann U, Gao Y, Pandey B, Klinge S, Warzecha K.-D, Krueger C, Roth HD, Demuth M. J. Am. Chem. Soc. 1993; 115: 10358
- 52b Heinemann C, Demuth M. J. Am. Chem. Soc. 1997; 119: 1129
- 52c Heinemann C, Demuth M. J. Am. Chem. Soc. 1999; 121: 4894
- 52d Goeller F, Heinemann C, Demuth M. Synthesis 2001; 1114
- 52e Rosales V, Zambrano J, Demuth M. Eur. J. Org. Chem. 2004; 1798
- 52f Ozser ME, Icil H, Makhynya Y, Demuth M. Eur. J. Org. Chem. 2004; 3686
- 53 Yang Z, Li H, Zhang L, Zhang M.-T, Cheng J.-P, Luo S. Chem. Eur. J. 2015; 21: 14723
- 54 Yang Z, Li S, Luo S. Acta Chim. Sin. 2017; 75: 351
- 55 Trotta AH. J. Org. Chem. 2017; 82: 13500
For recent reviews in this area, see