
Subscribe to RSS
DOI: 10.1055/a-2526-0771
Recent Advances in the Synthesis of Functionalized Pyrazolo[1,5-a]pyrimidines via C–H Functionalization
A.K.B. acknowledges the Department of Science and Technology and Biotechnology (DSTBT), Government of West Bengal, India (GO no. 324(Sanc.)/STBT-11012(25)/13/2024-ST SEC) for financial support. A.K.B. also acknowledges the financial support from the SERB, DST (File no. EEQ/2018/000498) and the University of Kalyani (PRG). S. P. (CSIR-SRF), T. C. (URS-SRF) and S. D. (UGC-SRF) acknowledge the Council of Scientific and Industrial Research (CSIR) New Delhi, University of Kalyani, and University Grants Commission (UGC), New Delhi, respectively, for their fellowships.

This article is dedicated to Prof. B. C. Ranu on the grand occasion of his 75th birthday
Abstract
Recent developments in the synthesis of functionalized pyrazolo[1,5-a]pyrimidines through C–H functionalization have been summarized in this account, covering the synthesis of 3-halo, 3-nitro, 3-formyl, 3-acetyl, 3-sulfenyl, 3-selenyl, and 3-thiocyanato pyrazolo[1,5-a]pyrimidines and bis(pyrazolo[1,5-a]pyrimidin-3-yl)methanes. The main focus highlights the utilization of sustainable conditions in designing the protocols. Mechanistic aspects of these protocols have also been discussed in detail.
1 Introduction
2 Discussion
2.1 Synthesis of 3-Halo Pyrazolo[1,5-a]pyrimidines
2.2 Synthesis of 3-Nitro Pyrazolo[1,5-a]pyrimidines
2.3 Synthesis of 3-Formyl Pyrazolo[1,5-a]pyrimidine
2.4 Synthesis of 3-Acetyl Pyrazolo[1,5-a]pyrimidines
2.5 Synthesis of 3-Sulfenyl Pyrazolo[1,5-a]pyrimidines
2.6 Synthesis of 3-Selenyl Pyrazolo[1,5-a]pyrimidines
2.7 Synthesis of 3-Thiocyanated Pyrazolo[1,5-a]pyrimidines
2.8 Synthesis of Bis(pyrazolo[1,5-a]pyrimidinyl)methanes
3 Conclusions and Outcome
Key words
pyrazolo[1,5-a]pyrimidines - C–H functionalization - photocatalysis - electrocatalysis - green chemistryBiographical Sketches

Suvam Paul was born in Berhampore, Murshidabad. He completed B.Sc. degree in chemistry in 2018 from Sripat Singh College, affiliated to the University of Kalyani. He completed his M.Sc. degree in 2020 from the University of Kalyani. He qualified CSIR-NET November 2020 with a JRF. Now he is currently pursuing a Ph.D. as a CSIR-SRF in chemistry at the University of Kalyani. His research focuses on the exploration of visible-light photocatalysis in the synthesis of heterocycles.

Tathagata Choudhuri was born in Purba Bardhaman, West Bengal, India. He graduated in chemistry in 2018 from Krishnath College, Berhampore, Murshidabad. He completed his M.Sc. in chemistry in 2020 from the University of Kalyani, Kalyani, Nadia. He is currently pursuing his Ph.D. as a URS-SRF at the University of Kalyani, Nadia, West Bengal, India under the supervision of Dr. Avik Kumar Bagdi. His research focuses on the development of new metal-free methodologies for synthesizing heterocycles.

Sourav Das was born in July 1996 in the district of Murshidabad, WB, India. He completed his B.Sc. in 2017 from Sripat Singh College, Jiaganj, WB affiliated to the University of Kalyani and M.Sc. in 2019 from the University of Kalyani. He is currently working as a UGC-SRF under the supervision of Dr. Avik Kumar Bagdi at the University of Kalyani. His research focuses on synthesis of novel heterocycles through oxidative cyclization.

Papiya Sikdar was born in February 1992 at Gopalnagar in the district of North 24 Parganas, WB, India. She completed her B.Sc. in 2013 from Dinabandhu Mahavidyalaya, Bongaon, WB affiliated to WBSU and M.Sc. in 2015 from West Bengal State University, Barasat. Currently she is pursuing her Ph.D. at the University of Kalyani under the supervision of Dr. Avik Kumar Bagdi. Her research focuses on the synthesis of heterocycles through oxidative C–H functionalization.

Dr. Avik Kumar Bagdi obtained his Ph.D. from the Visva-Bharati, India in 2014 under the guidance of Dr. Alakananda Hajra. He received 2nd Prize of ‘2014 Eli Lilly & Company Asia Outstanding Thesis Award’. In 2015, he was appointed as an Assistant Professor at Triveni Devi Bhalotia College, Raniganj. In 2016, he went to OIST, Japan to carry out his Post-Doctoral Research with Prof. Fujie Tanaka. Since 2018, Dr. Bagdi has worked as an Assistant Professor in the Department of Chemistry, University of Kalyani, India. His current research interests include the employment of visible light photocatalysis in the synthesis and functionalization of bioactive heterocycles.
Introduction
Pyrazolo[1,5-a]pyrimidine, a nitrogen-containing 5,6-fused heterocycle, is frequently found in various commercially available pharmaceuticals (Figure [1]).[1] Zaleplon (1), Indiplon (2), Lorediplon (3), and Ocinaplon (4) are nonbenzodiazepine hypnotic drugs whereas Reversan (5), Repotrectinib (6), Larotrectinib (7), Selitrectinib (8), Dinaciclib (9), and Dorsomorphin (10) are useful anticancer drugs.[2] On the other hand, Presatovir (11) is an antiviral drug and Anagliptin (12) is an antidiabetic drug.[3] Shard and co-workers demonstrated the antiproliferative effects of pyrazolo[1,5-a]pyrimidine with a ferrocene moiety against oral cancer.[4] All these pharmaceuticals contain the pyrazolo[1,5-a]pyrimidine scaffold as a core structure. This scaffold is also found in agrochemicals like pyrazophos (13) (Figure [2]).[5] Different pyrazolo[1,5-a]pyrimidines have also attracted the interest of material chemists due to their exciting optical properties and usefulness in sensing different analytes.[6] The Portilla group exemplified the usefulness of pyrazolo[1,5-a]pyrimidine-based salts 14 and 15 for cyanide sensing (Figure [3]).[7] They demonstrated that pyrazolo[1,5-a]pyrimidine derivative 16 could be used to quantify ethanol content in hydrocarbons and distilled spirits.[8] They also showed that pyrazolo[1,5-a]pyrimidine derivative 17 is highly useful in the quantification of water in organic solvents.[9] So, it is highly important to have synthetic methodologies for different functionalized pyrazolo[1,5-a]pyrimidine derivatives.
Heterocycles are the core structure of most of the drug molecules.[10] Currently, the design of ‘green’ synthetic strategies for heterocycles is important in terms of improved sustainability and resource-economy.[11] In this context, strategies involving C–H functionalization have attracted the attention of synthetic chemists.[12] No pre-functionalization of the substrate is required for C–H functionalization. Different sustainable catalysts have been employed for the synthesis of functionalized heterocycles through C–H functionalization.[13] These include iodine/hypervalent iodine catalysis, photocatalysis, electrocatalysis, and transition metal catalysis. Microwave irradiation and ultrasonication have been utilized for C–H functionalization.[14]



Different functionalized pyrazolo[1,5-a]pyrimidine derivatives have been synthesized over the years.[15] C–H functionalization has been also employed in the synthesis of these derivatives (Scheme [1]). The importance of pyrazolo[1,5-a]pyrimidines in different fields prompted us to develop new synthetic methodologies for this scaffold. Our main focus was to design new strategies for synthesizing functionalized pyrazolo[1,5-a]pyrimidines through C–H functionalization under environmentally friendly conditions. In this account, we will discuss our efforts in this area. Recent reports on this topic from other groups will also be highlighted (2015–2024).

Discussion
2.1Synthesis of 3-Halo Pyrazolo[1,5-a]pyrimidines
3-Halo pyrazolo[1,5-a]pyrimidines are useful synthetic intermediates for the synthesis of different pyrazolo[1,5-a]pyrimidines and also show anxiolytic properties.[16] In 2017, the Portilla group developed an efficient strategy to synthesize 3-halo pyrazolo[1,5-a]pyrimidines via one-pot cyclization followed by C–H halogenation (Scheme [2]).[17] Reaction between amino pyrazoles 18 and β-enaminones 19 under microwave conditions at 180 °C generated pyrazolo[1,5-a]pyrimidine derivatives 20. Subsequent aromatic electrophilic substitution reaction of these derivatives with N-halosuccinimides (NXS) as halogenating agents in 1,2-dichloroethane (DCE) at room temperature afforded 3-halo pyrazolo[1,5-a]pyrimidines 21. This one-pot two-step protocol was applied towards various pyrazoles and β-enaminones and halogenated pyrazolo[1,5-a]pyrimidines were synthesized in excellent yields. The protocol was well suited to the iodination, bromination, and chlorination of pyrazolo[1,5-a]pyrimidines using NXS (X = Cl, Br, I).
In 2023, our research group developed a tandem one-pot cyclization and oxidative halogenation strategy for the synthesis of 3-halo pyrazolo[1,5-a]pyrimidines 21 using NaX (X = Cl, Br, I) as the halogenating agents (Scheme [3]).[18] This strategy is useful for synthesizing iodinated pyrazolo[1,5-a]pyrimidines employing amino pyrazoles 18, β-enaminones, and NaI in one step with the aid of K2S2O8 as an oxidant. K2S2O8 was best among various oxidants, such as PIDA, K2S2O8, Na2S2O8, and (NH4)2S2O8, and water was the best solvent among MeCN, DCE, MeOH, EtOH, DMF, and water. Various amino pyrazoles and β-enaminones were treated with sodium iodide in this three-component reaction. β-Enaminones containing electron-donating and electron-withdrawing groups afforded the desired 3-iodo pyrazolo[1,5-a]pyrimidines in good to excellent yields. The methodology was applicable to the synthesis of 3-bromo/chloro pyrazolo[1,5-a]pyrimidines using sodium bromide/chloride, however, the reaction was executed in a one-pot, two-step manner. This strategy afforded the desired brominated product in good yields (72–81%) whereas a 3-chloro pyrazolo[1,5-a]pyrimidine was obtained in 57% yield. This one-pot strategy was applied to (E)-chalcone (22) instead of β-enaminones (Scheme [3]). However, water was not fruitful for this transformation and DMSO was proven to be the best solvent in this case. Various 3-iodo pyrazolo[1,5-a]pyrimidines were obtained in good yields through this one-pot, three-component reaction of amino pyrazoles, chalcone, and NaI.


The reaction was extended to the oxidative C–H halogenations of pyrazolo[1,5-a]pyrimidines employing NaX as halogenating agents and K2S2O8 as an oxidant (Scheme [4]). Pyrazolo[1,5-a]pyrimidines 20 with electron-donating and electron-withdrawing functionalities, like -Me, -OMe, -F, -Cl, -Br, -NO2, etc., gave 3-halo pyrazolo[1,5-a]pyrimidines in excellent yields. This C–H halogenation strategy offered much better yields over the one-pot approach. Importantly, regioselective iodination/bromination occurred only on the pyrazole ring in the case of pyrazolo[1,5-a]pyrimidines with heteroaryl moieties.

The reaction proceeds through a non-radical pathway as the iodination reaction was not quenched in the presence of radical scavengers (TEMPO or 1,1-diphenylethylene (DPE)). Additionally, the direct C–H halogenation of pyrazolo[1,5-a]pyrimidines was also possible to obtain 3-halo pyrazolo[1,5-a]pyrimidines. Thus the reaction proceeds through initial formation of pyrazolo[1,5-a]pyrimidine derivatives. Accordingly, the proposed mechanism of this tandem cyclization-C–H halogenation is outlined in Scheme [5]. Michael addition of amino pyrazole to the β-enaminone/chalcone followed by intramolecular cyclization affords a pyrazolo[1,5-a]pyrimidine derivative 20 that undergoes an aromatic electrophilic substitution reaction with in situ generated I2 to produce the 3-iodo pyrazolo[1,5-a]pyrimidine.

The usefulness of 3-halo pyrazolo[1,5-a]pyrimidine as a synthetic building block in synthesizing highly substituted pyrazolo[1,5-a]pyrimidines 26 and 27 via cross-coupling reactions was also demonstrated (Scheme [6]). Wide substrate scopes, gram-scalability, regioselectivity, and use of water as a solvent are the important features of this protocol.

In 2024, the Kshirsagar group reported oxidant-mediated C–H halogenations of pyrazolo[1,5-a]pyrimidines using KX (X = Cl, Br, I) as halogenating agents at room temperature (Scheme [7]).[19] Optimization of the reaction conditions showed phenyliodine diacetate (PIDA) to be the best oxidant for the halogenation among various oxidants, such as K2S2O8, Na2S2O8, oxone, TBPB, TBHP, PIFA, and PIDA. The optimum yield was achieved when the reaction was carried out using 1.5 equiv. KI, and 1 equiv. PIDA in water. This strategy was applied to the halogenation of various substituted pyrazolo[1,5-a]pyrimidines with -Me, -OMe, -F, -Cl, and -Br functionalities. This strategy was well suited for iodination, bromination, and chlorination of pyrazolo[1,5-a]pyrimidines. The use of MeOH as solvent instead water in the case of chlorination was found to be effective for better yields. Simple pyrazolo[1,5-a]pyrimidine also reacted well to afford regioselectively 3-halo pyrazolo[1,5-a]pyrimidines. They demonstrated the gram-scale applicability of the methodology and also utilized the synthesized 3-iodo pyrazolo[1,5-a]pyrimidines in the preparation of highly substituted pyrazolo[1,5-a]pyrimidines through cross-coupling reactions.

The proposed mechanism is presented in Scheme [8]. Initially, a ligand exchange reaction happens between PIDA and metal halides which forms intermediate 28. This intermediate 28 is converted into a hypohalite salt AcOX 29 by eliminating iodobenzene. This hypohalite salt AcOX 29 serves as an electrophilic halogen source and on reaction with pyrazolo[1,5-a]pyrimidines generates another intermediate 30. This intermediate 30 is converted into the 3-halo pyrazolo[1,5-a]pyrimidine after deprotonation.


At the same time, our group also developed a visible-light-promoted photocatalyst-free iodination strategy of pyrazolo[1,5-a]pyrimidines (Scheme [9]).[20] PIDA acted as an iodinating agent in the presence of p-toluenesulfonic acid (PTSA) under the irradiation of visible light. PTSA was proven to be the best additive among PTSA, TFA, AcOH, benzoic acid, formic acid, and NaOAc. Whereas among various solvents, ethanol showed the best result in affording the iodinated product. The use of molecular iodine instead of PIDA exhibited a detrimental effect as a much lower yield was obtained. Visible light was indispensable in this strategy as no product was formed under dark conditions whereas only 22% yield was observed when it was carried out at 60 °C under dark conditions. The utilization of the optimized reaction conditions towards various pyrazolo[1,5-a]pyrimidine derivatives revealed the wide applicability of the methodology. Both electron-donating and electron-withdrawing groups, like -Me, - t Bu, -OMe, -F, -Cl, -Br, -CN, -NO2, etc., were well tolerated. Pyrazolo[1,5-a]pyrimidine derivatives with heteroaryl moieties also afforded the iodinated pyrazolo[1,5-a]pyrimidines and iodination took place only on the pyrazole ring.
The practical applicability was demonstrated by carrying out a gram-scale preparation of 3-iodo pyrazolo[1,5-a]pyrimidines (Scheme [10]). Importantly, the methodology could easily be executed under the irradiation of sunlight. Lastly, a synthesized 3-iodo pyrazolo[1,5-a]pyrimidine was converted into other functionalized pyrazolo[1,5-a]pyrimidines 27, 32–35 through arylation, alkenylation, alkynylation, sulfenylation, and selenylation reactions.

A trace amount of product was observed in the presence of radical scavengers such as TEMPO, BHT, and DPE, which supports the involvement of the radical pathway in this reaction. On the other hand, from the results of UV-vis spectra, it was assumed that an EDA complex is generated between pyrazolo[1,5-a]pyrimidine and PIDA in the presence of PTSA under the irradiation of blue LEDs. The proposed mechanism based on these results is outlined in Scheme [11]. Initially, a diradical species 36 is generated on reaction of pyrazolo[1,5-a]pyrimidine with PIDA under the irradiation of blue LEDs. This diradical species 36 is converted into another intermediate 37. The intermediate 37 is converted into the final product after reductive elimination.
Synthesis of 3-Nitro Pyrazolo[1,5-a]pyrimidines
The Portilla group also extended their methodology for the halogenations of pyrazolo[1,5-a]pyrimidines towards the synthesis of 3-nitro pyrazolo[1,5-a]pyrimidines 38.[17] They demonstrated the microwave-assisted synthesis of 3-nitro pyrazolo[1,5-a]pyrimidines through the cyclization between amino pyrazoles and β-enaminones followed by nitration by using HNO3/H2SO4 (2:1) as nitrating reagent (Scheme [12]). Microwave irradiation was essential for both of these steps. They also synthesized 3-amino pyrazolo[1,5-a]pyrimidines 39 through the reduction of the synthesized 3-nitro pyrazolo[1,5-a]pyrimidines employing 10% Pd/C-PPh3-CuI and Et3N under microwave irradiation.


Synthesis of 3-Formyl Pyrazolo[1,5-a]pyrimidines
Inspired by their previous results, the Portilla group in 2018 synthesized 3-formyl pyrazolo[1,5-a]pyrimidines 40 through a one-pot cyclo-condensation reaction followed by Vilsmeier–Haack formylation reaction under microwave conditions (Scheme [13]).[21] The first step was the cyclo-condensation reaction of amino pyrazoles with β-enaminones 18 whereas the second step was the microwave-assisted formylation reaction of 20 using POCl3/DMF as the formylating agent. This microwave-assisted one-pot methodology was applied to a series of β-enaminones and amino pyrazoles and in all cases good to excellent yields were obtained. The presence of an alkyl or aryl moiety in the 2-position of the pyrazole ring did not alter the reaction. Heteroaryl β-enaminones with pyridyl and thienyl moieties also afforded the formylated product in good yields. 3-Formyl pyrazolo[1,5-a]pyrimidine derivatives were further transformed into other functionalized pyrazolo[1,5-a]pyrimidines 41–43 through functional group interconversion. They also highlighted the photophysical properties of the synthesized 3-formyl pyrazolo[1,5-a]pyrimidine derivatives.

Synthesis of 3-Acetyl Pyrazolo[1,5-a]pyrimidines
Inspired by the results of the formylation reaction, the Portilla group in 2022 reported a microwave-assisted acetylation reaction of pyrazolo[1,5-a]pyrimidines utilizing acetic anhydride as an acetylating reagent (Scheme [14]).[22] They observed that the optimum yield could be obtained by carrying out the reaction in the presence of BF3·OEt2 (10 equiv.) in acetic acid. Other anhydrides, such as butanoic, trifluoroacetic, chloroacetic and phthalic anhydride, were not effective for this transformation. A much lower yield was observed with other solvents such as acetonitrile, DMF, DCE, THF, dioxane, and CS2. After achieving the standard reaction conditions, the authors employed various pyrazolo[1,5-a]pyrimidines to show the applicability of this methodology. Various electron-rich, electron-deficient, and sterically hindered pyrazolo[1,5-a]pyrimidines were well tolerated and provided 3-acetyl pyrazolo[1,5-a]pyrimidines 44 in excellent yields within a short reaction time. They also studied the photophysical properties of the synthesized 3-acetyl pyrazolo[1,5-a]pyrimidines and observed that the presence of an aryl moiety at the 5- and 7-position of the pyrazolo[1,5-a]pyrimidine scaffold has a significant effect on the photophysical properties.

Synthesis of 3-Sulfenyl Pyrazolo[1,5-a]pyrimidines
In 2023, our group developed a strategy for the regioselective 3-sulfenylation of pyrazolo[1,5-a]pyrimidines with thiols via cross-dehydrogenative coupling under blue LED irradiation (Scheme [15]).[23] Optimization of the reaction showed that the presence of Rose Bengal (RB) as a photocatalyst, KI as an additive, and K2S2O8 as an oxidant were necessary for this transformation under irradiation by visible light. The optimum formation of 3-sulfenyl pyrazolo[1,5-a]pyrimidine was observed in the presence of 2 mol% Rose Bengal, 20 mol% KI, and 1 equiv. K2S2O8 under irradiation by blue LEDs for 6 h. Light is indispensable as in the absence of this the reaction did not proceed. The reaction did not proceed under an inert atmosphere, which showed the crucial role of oxygen for the conversion of the product. Then this strategy was applied to a series of pyrazolo[1,5-a]pyrimidine derivatives containing different electron-donating and electron-withdrawing substituents. In all cases, 3-sulfenylated pyrazolo[1,5-a]pyrimidines were observed in good to excellent yields. 2-Furyl- and 2-pyridyl-substituted pyrazolo[1,5-a]pyrimidines also afforded the 3-sulfenylated products in 82% and 79% yield, respectively. 2,5,7-Trisubstituted pyrazolo[1,5-a]pyrimidines were also tolerated under this methodology. This methodology was also compatible with various thiophenol derivatives. Thiophenols containing electron-donating groups, such as -Me, -OMe, and electron-withdrawing groups, like -CF3 and -COOMe, afforded the desired products in good yields. ortho-Substituted thiophenols also reacted under the optimized condition. Pyridine-2-thiol and benzothiazole-2-thiol also reacted well however alkanethiols were not effective coupling partners for the sulfenylation reaction. Importantly, the sulfenylation took place regioselectively at the 3-position of the pyrazolo[1,5-a]pyrimidine.

Many experiments were performed to obtain details about the reaction mechanism. The reaction was totally quenched in the presence of singlet oxygen quencher DABCO and radical scavenger TEMPO. These results suggested the involvement of singlet oxygen and radical intermediate. The formation of the thiyl radical was detected using HRMS by generating its adduct with TEMPO. The formation of diphenyl disulfide was observed when thiophenol was treated with Rose Bengal under irradiation by blue LEDs. Based on the results of controlled experiments a plausible mechanism is suggested as shown in Scheme [16]. Initially, RB is excited to RB* under irradiation by blue LEDs. The excited state RB* converts the triplet oxygen (3O2) into singlet oxygen (1O2) via an energy transfer process and regenerates ground state RB. On the other hand, RB* also accepts an electron from thiophenol generating RB•− and radical cation intermediate 47. This RB•− is then converted into RB by converting 1O2 to O2 •−. O2 •− then converts 47 into the thiyl radical 48 by proton abstraction. Thiyl radical 48 is converted into diphenyl disulfide intermediate 49. An iodine radical is generated by the reaction of iodide with persulfate under blue LED irradiation. This iodine radical converts intermediate 49 into 50 and ArSI through reaction with pyrazolo[1,5-a]pyrimidine. Then intermediate 50 is converted into the product either by hydrogen abstraction by HO2 • or by persulfate-mediated oxidation followed by proton abstraction. ArSI on reaction with pyrazolo[1,5-a]pyrimidine gives the final product by electrophilic aromatic substitution.

The methodology is well-suited for the gram-scale preparation of 3-sulfenylated pyrazolo[1,5-a]pyrimidine derivatives and could be executed under sunlight (Scheme [17]). A 3-sulfenylated pyrazolo[1,5-a]pyrimidine derivative was easily oxidized to 3-sulfonyl pyrazolo[1,5-a]pyrimidine derivative 52 by oxidation with mCPBA.

In 2024, the Kshirsagar group developed a new electrochemical strategy for the sulfenylation of pyrazolo[1,5-a]pyrimidines (Scheme [18]).[24] The reaction was carried out using diphenyl disulfide as sulfenylating reagent and 20 mol% TBABF4 as an electrolyte with HCl as an additive in CH3CN under galvanostatic conditions. Acetonitrile was proven to be the best solvent among DMSO, DMF, and MeOH. The use of other acids as an additive, such as AcOH, TFA, and H2SO4, was not as effective as HCl in affording the sulfenylated product. No product was detected without electricity. Even decreasing the amount of current decreased the yields of the sulfenylated product. This standard optimized methodology was then applied to various pyrazolo[1,5-a]pyrimidines. In all cases, the regioselective formation of 3-sulfenylated pyrazolo[1,5-a]pyrimidines 46 was observed in good yields.

Synthesis of 3-Selenyl Pyrazolo[1,5-a]pyrimidines
In 2023, our group developed an erythrosine B (EB) catalyzed regioselective selenylation of pyrazolo[1,5-a]pyrimidines using diaryl or dialkyl diselenides as the selenylating reagent under irradiation by visible light (Scheme [19]).[25] During optimization it was observed that erythrosine B acted as the best photocatalyst for this transformation among fluorescein, methylene blue, eosin Y, Na2eosin Y, and erythrosine B. A much lower yield was observed in the absence of any photocatalyst whereas no selenylated product was detected in the absence of light, which shows the importance of a light source for this strategy. Molecular oxygen played an important role in this transformation which increased the yield of the selenylated product. The optimum yield was achieved by carrying out the reaction using 3 mol% erythrosine B in MeOH under an oxygen atmosphere with irradiation by white LEDs for 24 h. The optimized reaction conditions were utilized to react various substituted pyrazolo[1,5-a]pyrimidines 20 with diselenides. 7-Phenyl-substituted pyrazolo[1,5-a]pyrimidines bearing electron-donating as well as electron-withdrawing groups in the phenyl ring afforded the desired selenylated products 54 in good to excellent yields. The presence of a 7-(1-naphthyl) group in the pyrazolo[1,5-a]pyrimidine gave the selenyl-substituted product in 77% yield, while a 7-(2-furyl) or 7-(2-pyridyl) group gave the selenylated product in 63% and 81% yield, respectively. The methodology is applicable to various diaryl diselenides, diheteroaryl diselenides, and dialkyl diselenides to obtain the respective selenylated pyrazolo[1,5-a]pyrimidines in good yields.

This methodology was then extended to 2,5,7-trisubstituted pyrazolo[1,5-a]pyrimidines (Scheme [20]). Pyrazolo[1,5-a]pyrimidines 20 substituted at C7 with Me-, MeO-, F-, Cl-, and Br-substituted phenyl rings gave the desired selenylated product in good yields which shows the efficiency of that methodology towards bulky substituents. 2,5,7-Triphenyl- and 2,5,7 trimethyl-substituted pyrazolo[1,5-a]pyrimidines afforded the selenylated products in 91% and 74% yield, respectively.

To get an insight into the mechanistic pathway, a detailed study was carried out. In the presence of TEMPO, the reaction did not fully quench. This result excluded the possibility of the radical pathway. When diphenyl diselenide reacted with DPE under standard conditions 2-methoxy-2,2-diphenylethyl phenyl selenide (55) was observed. This result suggested the formation of PhSe+ under the reaction medium. A plausible mechanism was drawn based on these experiments (Scheme [21]). EB is excited into EB* under irradiation by white LED and transfers its energy to diphenyl diselenide converting it into PhSe• and regenerating ground state EB. PhSe• on reaction with oxygen is converted into PhSe+ 57. Pyrazolo[1,5-a]pyrimidine then reacts with PhSe+ 57 to generate intermediate 58. Intermediate 58 then affords the selenylated product on hydrogen atom abstraction.

In the same year, our group reported a photocatalyst-free selenylation of pyrazolo[1,5-a]pyrimidines in the presence of K2S2O8 under the irradiation of 23-W white LEDs (Scheme [22]).[26] The reaction was optimized and the best conditions were using 1.2 equiv. of K2S2O8 in DMSO under irradiation by white LEDs for 20 h. Various oxidants were also screened, such as PIDA, TBHP, (NH4)2S2O8, Na2S2O8, and K2S2O8, with K2S2O8 giving the best results. Molecular oxygen was not suitable for this transformation as only 33% product was observed in this case. A much lower yield was observed in the absence of light, which showed the necessity of light for this reaction. After achieving the best conditions, this strategy was applied to various pyrazolo[1,5-a]pyrimidines. Differently substituted pyrazolo[1,5-a]pyrimidines 20 reacted well under these conditions and afforded the desired product 54 in good yields. Heteroaryl-containing pyrazolo[1,5-a]pyrimidines such as 2-pyridyl- and 2-furyl-pyrazolo[1,5-a]pyrimidines gave the corresponding selenylated products in 70% and 74% yields, respectively. This strategy was well suited for diaryl diselenide derivatives. Diphenyl selenides bearing Me, OMe, and Br groups afforded the products in 79%, 78%, and 75% yields, respectively.

The reaction proceeded through a radical pathway and the proposed mechanism is outlined in Scheme [23]. Initially, PhSe• is generated from (PhSe)2 under irradiation by white LEDs in the presence of K2S2O8. Then pyrazolo[1,5-a]pyrimidine reacts with PhSe• to generate an intermediate 59 which on further oxidation affords a cationic intermediate 58. Finally, the desired product is achieved by proton abstraction from intermediate 58.

In 2024, the Kshirsagar group developed the Oxone-mediated selenylation of pyrazolo[1,5-a]pyrimidines using 1 equiv. of Oxone as an oxidant and 1 equiv. of diphenyl diselenide in acetonitrile at room temperature (Scheme [24]).[27] Other oxidants like PIDA, K2S2O8, and Na2S2O8 were not as productive as Oxone. The authors utilized the standard reaction conditions towards various pyrazolo[1,5-a]pyrimidine derivatives to show the applicability of the protocol. Both electron-donating and electron-withdrawing groups containing pyrazolo[1,5-a]pyrimidines 20 reacted well under the standard conditions to provide the respective selenylated product 54 in excellent yields. This methodology was well suited for different diaryl diselenide derivatives. In all cases, excellent yields were obtained.
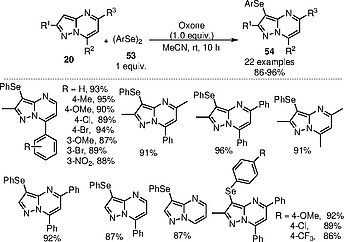
Based on the results of controlled experiments with radical scavengers like TEMPO and BHT, a mechanistic pathway was proposed as shown in Scheme [25]. Initially reaction of (PhSe)2 with Oxone generates an electrophilic chalcogen species 60. Attack by the pyrazolo[1,5-a]pyrimidines to this intermediate 60 at the more nucleophilic 3-position generates intermediate 58. Finally the 3-selenylated pyrazolo[1,5-a]pyrimidine is formed by deprotonation of intermediate 58.

In 2024, our group developed a one-pot strategy for the synthesis of selenylated pyrazolo[1,5-a]pyrimidines from readily available amino pyrazoles, chalcones or β-enaminones, and diselenides (Scheme [26]).[28] In this method, the extraction and purification of the pyrazolo[1,5-a]pyrimidines was not required. Molecular iodine was the most suitable catalyst and only 5 equiv. of DMSO was required for this transformation. CuI was less effective and FeCl3 was ineffective for this one-pot synthesis. The optimum yield was obtained by carrying out the reaction using 20 mol% I2 and 5 equiv. of DMSO at 110 °C. This standard methodology was then applied to various chalcone derivatives. Chalcones derivatives 22 with electron-donating as well as electron-withdrawing groups on both phenyl rings afforded the selenylated products 54 in good to excellent yields. It was observed that only one regioisomer was obtained in all cases. Heteroaryl enones also afforded the respective selenylated products in moderate yields. This methodology was also compatible with various diaryl diselenide derivatives. Electron-donating as well as electron-withdrawing substituents on the diphenyl diselenides also reacted well to give the selenylated products. Dialkyl diselenides like dibenzyl diselenides and dimethyl diselenides were also susceptible to this reaction conditions and afforded the desired products in moderate yields.

This method was also applicable to β-enaminone systems 19 instead of chalcones (Scheme [27]). This two-step one-pot approach is useful for the synthesis of 3-selenylated pyrazolo[1,5-a]pyrimidines from amino pyrazoles, β-enaminones, and diphenyl diselenide. Different β-enaminones 19 with electron-donating as well as electron-withdrawing substituents afforded selenylated pyrazolo[1,5-a]pyrimidines 54 in good to excellent yields. A heteroaryl β-enaminone containing a 2-pyridyl group was also suitable for this strategy affording the selenylated product in 69% yield.
The non-radical pathway of this reaction was proven on the basis of comparable yields of the reaction using TEMPO as a radical scavenger in comparison to those without TEMPO. The proposed mechanism of this three-component reaction is outlined in Scheme [28]. The reaction initiates with the formation of PhSeI from (PhSe)2 by the reaction of molecular iodine. 3-Selenylated pyrazolo[1,5-a]pyrimidines are formed through either initial cyclization of the amino pyrazole with chalcone to give the pyrazolo[1,5-a]pyrimidine followed by selenylation with PhSeI to give the selenyl pyrazolo[1,5-a]pyrimidine (Path I) or initial selenylation of the pyrazole with PhSeI followed by cyclization of the selenylated amino pyrazole 64 with chalcone (Path II). The rate of the first pathway (Path I) is much faster with respect to the second pathway (Path II).


In 2024, the Kshirsagar group demonstrated an electrochemical approach to the selenylation of pyrazolo[1,5-a]pyrimidine derivatives requiring no oxidant or catalyst for this C–H selenylation at rt (Scheme [29]).[24] Inexpensive graphite electrodes and 20 mol% TBABF4 as an electrolyte were utilized in this electrochemical reaction. The methodology was applied to a wide range of pyrazolo[1,5-a]pyrimidines with different functionalities, like -Me, -OMe, -F, -Cl, -Br, -I, -CN, etc., and towards various diaryl diselenides. Importantly, selenylation of pyrazolo[1,5-a]pyrimidine was also successful and gave regioselectively 3-(phenylselanyl)pyrazolo[1,5-a]pyrimidine in 88% yield. Dimethyl diselenide was also used successfully and gave the corresponding selenylated pyrazolo[1,5-a]pyrimidine derivatives.

Synthesis of 3-Thiocyanated Pyrazolo[1,5-a]pyrimidines
In 2019, the Petrosyan group developed a strategy for the thiocyanation of pyrazolo[1,5-a]pyrimidine via an electrochemical approach using NH4SCN as a thiocyanating reagent (Scheme [30]).[29] The reaction employed NH4SCN (3–4 equiv.) using platinum electrodes in the presence of 0.1 M NaClO4 as an electrolyte. Various 3-thiocyanato pyrazolo[1,5-a]pyrimidines 67 were successfully synthesized in 60–89% yield from pyrazolo[1,5-a]pyrimidines.

The Kshirsagar group modified this strategy for the C–H thiocyanation of pyrazolo[1,5-a]pyrimidines using Oxone as an oxidant and 2 equiv. of NH4SCN as the thiocyanating agent (Scheme [31]).[27] This thiocyanation reaction was employed various pyrazolo[1,5-a]pyrimidine derivatives using 1 equiv. Oxone to give thiocyanated pyrazolo[1,5-a]pyrimidines 67 in good to excellent yields.

In 2024, Chatterjee and co-workers also reported the C–H thiocyanation of pyrazolo[1,5-a]pyrimidines employing 2 equiv. of KSCN as the thiocyanating agent through a photochemical approach (Scheme [32]).[30] 9-Mesityl-10-methylacridinium perchlorate was the best photocatalyst for the thiocyanation of pyrazolo[1,5-a]pyrimidines in an oxygen atmosphere under irradiation by blue LEDs. The use of other photocatalysts, like eosin Y, Rose Bengal, 2,4,6-triphenylpyrylium tetrafluoroborate, fac-[Ir(ppy)3] salt, was not as effective as 9-mesityl-10-methylacridinium perchlorate. Oxygen played an important role, as the reaction in air gave only 45% yield whereas under an inert atmosphere the formation of a trace amount of product was observed. The methodology was applicable to various pyrazolo[1,5-a]pyrimidine derivatives with functionalities like -Me, -OMe, -F, -Cl, -Br, -CN, -NO2, -COOMe, -OH, etc. They found that higher yields were obtained in the case of pyrazolo[1,5-a]pyrimidines with electron-donating substituents compared to electron-withdrawing substituents. The reactions of naphthyl and heteroaryl (2-furyl and 2-thienyl) containing pyrazolo[1,5-a]pyrimidine derivatives were also successful, however, the yields were moderate. This strategy was extended to 2,5,7-trisubstituted pyrazolo[1,5-a]pyrimidines and gave highly substituted thiocyanated pyrazolo[1,5-a]pyrimidines in moderate to good yields.



The usefulness of thiocyanated pyrazolo[1,5-a]pyrimidine as a synthetic intermediate in the synthesis of other functionalized pyrazolo[1,5-a]pyrimidines was also shown (Scheme [33]). 2-Methyl-7-phenyl-3-thiocyanatopyrazolo[1,5-a]pyrimidine was treated with Cs2CO3 to give bis(2-methyl-7-phenylpyrazolo[1,5-a]pyrimidin-3-yl) disulfide (68). A click reaction of 2-methyl-7-phenyl-3-thiocyanatopyrazolo[1,5-a]pyrimidine with NaN3 gave 2-methyl-7-phenyl-3-[(1H-tetrazol-5-yl)thio]pyrazolo[1,5-a]pyrimidine (69).
A mechanistic pathway is proposed (Scheme [34]). Initially, the photocatalyst is excited under irradiation by blue LEDs. The excited state photocatalyst generates the SCN radical 70 by reaction with KSCN through a reductive quenching pathway. SCN radical 70 then reacts with pyrazolo[1,5-a]pyrimidine to generate intermediate 71. On the other hand, the reduced photocatalyst completes its catalytic cycle by reaction with oxygen thus generating the superoxide anion radical. This superoxide anion radical reacts with water to generate a hydroperoxide radical, which abstracts a hydrogen atom from intermediate 71 to generate the thiocyanated product.
Synthesis of Bis(pyrazolo[1,5-a]pyrimidinyl)methanes

In 2024, the Ma group developed a one-pot methodology for the synthesis of methylene-bridged bis(pyrazolo[1,5-a]pyrimidines) 74 using N,N-dimethylethanolamine (73) as a carbon synthon (Scheme [35]).[31] The [3+2+1] cycloaddition reaction of 3-amino pyrazoles 18, aryl ketones 72, and N,N-dimethylethanolamine (73) in the presence of FeCl3 as a Lewis acid at 140 °C gave bis(pyrazolo[1,5-a]pyrimidin-3-yl)methanes 74 in up to 84% yield. Other Lewis acids, like Cu(OTf)2, AlCl3, Zn(OAc)2, Sc(OTf)3, NiCl2, etc., were totally ineffective for this transformation. The rate of reaction was markedly increased in an oxygen atmosphere. Utilizing the optimum conditions, the scope of this methodology was explored using various ketones and amino pyrazoles. Aryl methyl ketones containing electron-donating and electron-withdrawing functionalities in either the ortho-, meta-, or para-position reacted smoothly to provide the corresponding products in moderate yields; heteroaryl methyl ketones also gave moderate yields. However, aliphatic ketones were not suitable substrates for this transformation. This methodology was examined using various 3-amino pyrazole derivatives. Amino pyrazoles containing electron-donating groups, such as -Me and -OMe, and electron-withdrawing groups, such as -Cl, and -Br, reacted smoothly to give the respective products. 3-Amino-5-tert-butyl-1H-pyrazole reacted with acetophenone and 73 to give the corresponding product in only 30% yield due to steric hindrance.

Also in 2024, the Chatterjee group reported the synthesis of methylene-bridged bis(pyrazolo[1,5-a]pyrimidines) employing DMSO as a methylene source under metal-free conditions (Scheme [36]).[32] The optimum yield was obtained by carrying out the reaction using Na2S2O5 (3 equiv.) as an oxidant and acetic acid as an additive at 120 °C for 16 h. The use of other acid additives, such as TFA, TsOH, and FeCl3, gave lower yields. This methodology was utilized with various pyrazolo[1,5-a]pyrimidines and it was found that unsubstituted pyrazolo[1,5-a]pyrimidine gave a moderate 48% yield while substituted pyrazolo[1,5-a]pyrimidines gave up to 90% yield. 2-Methyl-, 2-cyclopropyl-, and 2-phenyl-substituted pyrazolo[1,5-a]pyrimidines afforded the corresponding products in 85%, 67%, and 80% yield, respectively. 7-Phenyl-substituted pyrazolo[1,5-a]pyrimidines with various substitutions also reacted well to give the products in excellent yields.
Conclusions and Outcome
In summary, we have discussed the recent efforts towards the synthesis of functionalized pyrazolo[1,5-a]pyrimidine derivatives. The main objective was to highlight the methodologies involving C–H functionalization in the last 10 years (2015–2024). Different functional groups, such as halides, nitro, formyl, acetyl, sulfenyl, selenyl, thiocyanate, etc., were successfully incorporated at the 3-position of pyrazolo[1,5-a]pyrimidines. The usefulness of some synthesized pyrazolo[1,5-a]pyrimidines as synthetic building block has also been highlighted. Bis(pyrazolo[1,5-a]pyrimidin-3-yl)methanes were also synthesized through C–H functionalization. Several green techniques like the use of microwave irradiation, iodine catalysis, visible-light photocatalysis, and electrocatalysis have been efficiently employed for these synthetic protocols. All these methodologies exhibit several attractive features like the use of commercially available or easily accessible reagents, renewable energy resources, gram-scalability etc.
Although advancements in the synthesis of functionalized pyrazolo[1,5-a]pyrimidines have been made, these strategies have yet to be employed in the synthesis of natural products/pharmaceuticals. We believe this account will encourage the scientific community to explore this area. Additionally, this will be highly useful to synthetic chemists in the development of new functionalized pyrazolo[1,5-a]pyrimidine derivatives and other heterocycles.
Conflict of Interest
The authors declare no conflict of interest.
-
References
- 1 Zisapel N. Expert Opin. Invest. Drugs 2015; 24: 401
- 2a Arnold SL, Zhengming C, Phil S. US2008045547A1, 2008
- 2b Koilpillai JP, Kale SA, Kelkar LM, Zope SS, Khan MA. US2012028045A1, 2012
- 3a Mackman RL, Sangi M, Sperandio D, Parrish JP, Eisenberg E, Perron M, Hui H, Zhang L, Siegel D, Yang H, Saunders O, Boojamra C, Lee G, Samuel D, Babaoglu K, Carey A, Gilbert BE, Piedra PA, Strickley R, Iwata Q, Hayes J, Stray K, Kinkade A, Theodore D, Jordan R, Desai M, Cihlar T. J. Med. Chem. 2015; 58: 1630
- 3b Nishio S, Abe M, Ito H. Diabetes, Metab. Syndr. Obes.: Targets Ther. 2015; 18: 163
- 4 Gupta A, Das R, Chamoli A, Choithramani A, Kumar H, Patel S, Khude D, Bothra G, Wangdale K, Chowdhury MG, Rathod R, Mandoli A, Shard A. Organometallics 2022; 41: 2365
- 5 Ledieu MS, Helyer NL. Ann. Appl. Biol. 1983; 102: 275
- 6a Tigreros A, Aranzazu S.-L, Bravo N.-F, Zapata-Rivera J, Portilla J. RSC Adv. 2020; 10: 39542
- 6b Singsardar M, Sarkar R, Majhi K, Sinha S, Hajra A. ChemistrySelect 2018; 3: 1404
- 7 Tigreros A, Zapata-Rivera J, Portilla J. ACS Sustainable Chem. Eng. 2021; 9: 12058
- 8 Tigreros A, Macías M, Portilla J. Dyes Pigm. 2022; 202: 110299
- 9 Tigreros A, Macías M, Portilla J. ChemPhotoChem 2022; 6: e202200133
- 10 Eicher T, Hauptmann S, Speicher A. The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications . Wiley-VCH; Weinheim: 2003
- 11a Dhawa U, Kaplaneris N, Ackermann L. Org. Chem. Front. 2021; 8: 4886
- 11b Aricò F. Front. Chem. 2020; 8: 74
- 11c Constable DJ. C, Dunn PJ, Hayler JD, Humphrey GR, Leazer JL. Jr, Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007; 9: 411
- 12a Guillemard L, Kaplaneris N, Ackermann L, Johansson MJ. Nat. Rev. Chem. 2021; 5: 522
- 12b Pal T, Lahiri GK, Maiti D. Eur. J. Org. Chem. 2020; 2020: 6859
- 12c Davies HM. L, Morton D. J. Org. Chem. 2016; 81: 343
- 13a Yoshimura A, Zhdankin VV. Chem. Rev. 2024; 124: 11108
- 13b Bhanja R, Bera SK, Mal P. Adv. Synth. Catal. 2024; 366: 168
- 13c Mondal M, Ghosh S, Lai D, Hajra A. ChemSusChem 2024; 17: e202401114
- 13d Roy S, Panja S, Sahoo SR, Chatterjee S, Maiti D. Chem. Soc. Rev. 2023; 52: 2391
- 13e Baroliya PK, Dhaker M, Panja S, Al-Thabaiti SA, Albukhari SM, Alsulami QA, Dutta A, Maiti D. ChemSusChem 2023; 16: e202202201
- 13f Laskar R, Pal T, Bhattacharya T, Maiti S, Akita M, Maiti D. Green Chem. 2022; 24: 2296
- 13g Sinha SK, Guin S, Maiti S, Biswas JP, Porey S, Maiti D. Chem. Rev. 2022; 122: 5682
- 13h Bagdi AK, Rahman M, Bhattacherjee D, Zyryanov GV, Ghosh S, Chupakhin ON, Hajra A. Green Chem. 2020; 22: 6632
- 13i Parvatkar PT, Manetsch R, Banik BK. Chem. Asian J. 2019; 14: 6
- 14a Frecentese F, Sodano F, Corvino A, Schiano ME, Magli E, Albrizio S, Sparaco R, Andreozzi G, Nieddu M, Rimoli MG. Int. J. Mol. Sci. 2023; 24: 10722
- 14b Dhanush PC, Saranya PV, Anilkumar G. Tetrahedron 2022; 105: 132614
- 14c Yang H, Huang N, Wang N, Shen H, Teng F, Liu X, Jiang H, Tan M.-C, Gui Q.-W. ACS Omega 2021; 6: 25940
- 14d Besson T, Fruit C. Synthesis 2016; 48: 3879
- 15a Kumar H, Das R, Choithramani A, Gupta A, Khude D, Bothra G, Shard A. ChemistrySelect 2021; 6: 5807
- 15b Arias-Gómez A, Godoy A, Portilla J. Molecules 2021; 26: 2708
- 15c Salem MA, Helal MH, Gouda MA, Abd EL-Gawad HH, Shehab MA. M, El-Khalafawy A. Synth. Commun. 2019; 49: 1750
- 15d Cherukupalli S, Karpoormath R, Chandrasekaran B, Hampannavar GA, Thapliyal N, Palakollu VN. Eur. J. Med. Chem. 2017; 126: 298
- 16a Jismy B, Tikad A, Akssira M, Guillaumet G, Abarbri M. Molecules 2020; 25: 2062
- 16b Kirkpatrick WE, Okabe T, Hillyard IW, Robins RK, Dren AT, Novinson T. J. Med. Chem. 1977; 20: 386
- 17 Castillo J.-C, Rosero H.-A, Portilla J. RSC Adv. 2017; 7: 28483
- 18 Sikdar P, Choudhuri T, Paul S, Das S, Bagdi AK. ACS Omega 2023; 8: 23851
- 19 Chillal AS, Bhawale RT, Kshirsagar UA. RSC Adv. 2024; 14: 13095
- 20 Paul S, Das S, Choudhuri T, Sikdar P, Bagdi AK. Chem. Asian J. 2025; 20: e202401101
- 21a Castillo J.-C, Tigreros A, Portilla J. J. Org. Chem. 2018; 83: 10887
- 21b Correction: Castillo J.-C, Tigreros A, Cifuentes C, Portilla J. J. Org. Chem. 2024; 89: 14606
- 22 Aranzazu S.-L, Tigreros A, Arias-Gómez A, Zapata-Rivera J, Portilla J. J. Org. Chem. 2022; 87: 9839
- 23 Paul S, Das S, Choudhuri T, Sikdar P, Bagdi AK. J. Org. Chem. 2023; 88: 4187
- 24 Chillal AS, Bhawale RT, Sharma S, Kshirsagar UA. J. Org. Chem. 2024; 89: 14496
- 25 Choudhuri T, Paul S, Das S, Pathak DD, Bagdi AK. J. Org. Chem. 2023; 88: 8992
- 26 Sikdar P, Choudhuri T, Paul S, Das S, Kumar A, Bagdi AK. Synthesis 2023; 55: 3693
- 27 Chillal AS, Bhawale RT, Kshirsagar UA. ChemistrySelect 2024; 9: e202304815
- 28 Choudhuri T, Paul S, Sikdar P, Das S, Sawant SD, Bagdi AK. New J. Chem. 2024; 48: 9480
- 29 Kokorekin VA, Yaubasarova RR, Neverov SV, Petrosyan VA. Eur. J. Org. Chem. 2019; 2019: 4233
- 30 Pattanayak P, Satyanarayana AN. V, Chatterjee T. J. Org. Chem. 2024; 89: 13215
- 31 Zhang X, Chen J, Chen R, Wang L, Ma Y. Adv. Synth. Catal. 2024; 366: 3591
- 32 Pattanayak P, Satyanarayana AN. V, Saha S, Keerthana HS, Naresh A, Girase YK, Chatterjee T. Synlett 2024; 35: 2465
Corresponding Author
Publication History
Received: 09 January 2025
Accepted: 27 January 2025
Accepted Manuscript online:
27 January 2025
Article published online:
24 April 2025
© 2025. Thieme. All rights reserved
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Zisapel N. Expert Opin. Invest. Drugs 2015; 24: 401
- 2a Arnold SL, Zhengming C, Phil S. US2008045547A1, 2008
- 2b Koilpillai JP, Kale SA, Kelkar LM, Zope SS, Khan MA. US2012028045A1, 2012
- 3a Mackman RL, Sangi M, Sperandio D, Parrish JP, Eisenberg E, Perron M, Hui H, Zhang L, Siegel D, Yang H, Saunders O, Boojamra C, Lee G, Samuel D, Babaoglu K, Carey A, Gilbert BE, Piedra PA, Strickley R, Iwata Q, Hayes J, Stray K, Kinkade A, Theodore D, Jordan R, Desai M, Cihlar T. J. Med. Chem. 2015; 58: 1630
- 3b Nishio S, Abe M, Ito H. Diabetes, Metab. Syndr. Obes.: Targets Ther. 2015; 18: 163
- 4 Gupta A, Das R, Chamoli A, Choithramani A, Kumar H, Patel S, Khude D, Bothra G, Wangdale K, Chowdhury MG, Rathod R, Mandoli A, Shard A. Organometallics 2022; 41: 2365
- 5 Ledieu MS, Helyer NL. Ann. Appl. Biol. 1983; 102: 275
- 6a Tigreros A, Aranzazu S.-L, Bravo N.-F, Zapata-Rivera J, Portilla J. RSC Adv. 2020; 10: 39542
- 6b Singsardar M, Sarkar R, Majhi K, Sinha S, Hajra A. ChemistrySelect 2018; 3: 1404
- 7 Tigreros A, Zapata-Rivera J, Portilla J. ACS Sustainable Chem. Eng. 2021; 9: 12058
- 8 Tigreros A, Macías M, Portilla J. Dyes Pigm. 2022; 202: 110299
- 9 Tigreros A, Macías M, Portilla J. ChemPhotoChem 2022; 6: e202200133
- 10 Eicher T, Hauptmann S, Speicher A. The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications . Wiley-VCH; Weinheim: 2003
- 11a Dhawa U, Kaplaneris N, Ackermann L. Org. Chem. Front. 2021; 8: 4886
- 11b Aricò F. Front. Chem. 2020; 8: 74
- 11c Constable DJ. C, Dunn PJ, Hayler JD, Humphrey GR, Leazer JL. Jr, Linderman RJ, Lorenz K, Manley J, Pearlman BA, Wells A, Zaks A, Zhang TY. Green Chem. 2007; 9: 411
- 12a Guillemard L, Kaplaneris N, Ackermann L, Johansson MJ. Nat. Rev. Chem. 2021; 5: 522
- 12b Pal T, Lahiri GK, Maiti D. Eur. J. Org. Chem. 2020; 2020: 6859
- 12c Davies HM. L, Morton D. J. Org. Chem. 2016; 81: 343
- 13a Yoshimura A, Zhdankin VV. Chem. Rev. 2024; 124: 11108
- 13b Bhanja R, Bera SK, Mal P. Adv. Synth. Catal. 2024; 366: 168
- 13c Mondal M, Ghosh S, Lai D, Hajra A. ChemSusChem 2024; 17: e202401114
- 13d Roy S, Panja S, Sahoo SR, Chatterjee S, Maiti D. Chem. Soc. Rev. 2023; 52: 2391
- 13e Baroliya PK, Dhaker M, Panja S, Al-Thabaiti SA, Albukhari SM, Alsulami QA, Dutta A, Maiti D. ChemSusChem 2023; 16: e202202201
- 13f Laskar R, Pal T, Bhattacharya T, Maiti S, Akita M, Maiti D. Green Chem. 2022; 24: 2296
- 13g Sinha SK, Guin S, Maiti S, Biswas JP, Porey S, Maiti D. Chem. Rev. 2022; 122: 5682
- 13h Bagdi AK, Rahman M, Bhattacherjee D, Zyryanov GV, Ghosh S, Chupakhin ON, Hajra A. Green Chem. 2020; 22: 6632
- 13i Parvatkar PT, Manetsch R, Banik BK. Chem. Asian J. 2019; 14: 6
- 14a Frecentese F, Sodano F, Corvino A, Schiano ME, Magli E, Albrizio S, Sparaco R, Andreozzi G, Nieddu M, Rimoli MG. Int. J. Mol. Sci. 2023; 24: 10722
- 14b Dhanush PC, Saranya PV, Anilkumar G. Tetrahedron 2022; 105: 132614
- 14c Yang H, Huang N, Wang N, Shen H, Teng F, Liu X, Jiang H, Tan M.-C, Gui Q.-W. ACS Omega 2021; 6: 25940
- 14d Besson T, Fruit C. Synthesis 2016; 48: 3879
- 15a Kumar H, Das R, Choithramani A, Gupta A, Khude D, Bothra G, Shard A. ChemistrySelect 2021; 6: 5807
- 15b Arias-Gómez A, Godoy A, Portilla J. Molecules 2021; 26: 2708
- 15c Salem MA, Helal MH, Gouda MA, Abd EL-Gawad HH, Shehab MA. M, El-Khalafawy A. Synth. Commun. 2019; 49: 1750
- 15d Cherukupalli S, Karpoormath R, Chandrasekaran B, Hampannavar GA, Thapliyal N, Palakollu VN. Eur. J. Med. Chem. 2017; 126: 298
- 16a Jismy B, Tikad A, Akssira M, Guillaumet G, Abarbri M. Molecules 2020; 25: 2062
- 16b Kirkpatrick WE, Okabe T, Hillyard IW, Robins RK, Dren AT, Novinson T. J. Med. Chem. 1977; 20: 386
- 17 Castillo J.-C, Rosero H.-A, Portilla J. RSC Adv. 2017; 7: 28483
- 18 Sikdar P, Choudhuri T, Paul S, Das S, Bagdi AK. ACS Omega 2023; 8: 23851
- 19 Chillal AS, Bhawale RT, Kshirsagar UA. RSC Adv. 2024; 14: 13095
- 20 Paul S, Das S, Choudhuri T, Sikdar P, Bagdi AK. Chem. Asian J. 2025; 20: e202401101
- 21a Castillo J.-C, Tigreros A, Portilla J. J. Org. Chem. 2018; 83: 10887
- 21b Correction: Castillo J.-C, Tigreros A, Cifuentes C, Portilla J. J. Org. Chem. 2024; 89: 14606
- 22 Aranzazu S.-L, Tigreros A, Arias-Gómez A, Zapata-Rivera J, Portilla J. J. Org. Chem. 2022; 87: 9839
- 23 Paul S, Das S, Choudhuri T, Sikdar P, Bagdi AK. J. Org. Chem. 2023; 88: 4187
- 24 Chillal AS, Bhawale RT, Sharma S, Kshirsagar UA. J. Org. Chem. 2024; 89: 14496
- 25 Choudhuri T, Paul S, Das S, Pathak DD, Bagdi AK. J. Org. Chem. 2023; 88: 8992
- 26 Sikdar P, Choudhuri T, Paul S, Das S, Kumar A, Bagdi AK. Synthesis 2023; 55: 3693
- 27 Chillal AS, Bhawale RT, Kshirsagar UA. ChemistrySelect 2024; 9: e202304815
- 28 Choudhuri T, Paul S, Sikdar P, Das S, Sawant SD, Bagdi AK. New J. Chem. 2024; 48: 9480
- 29 Kokorekin VA, Yaubasarova RR, Neverov SV, Petrosyan VA. Eur. J. Org. Chem. 2019; 2019: 4233
- 30 Pattanayak P, Satyanarayana AN. V, Chatterjee T. J. Org. Chem. 2024; 89: 13215
- 31 Zhang X, Chen J, Chen R, Wang L, Ma Y. Adv. Synth. Catal. 2024; 366: 3591
- 32 Pattanayak P, Satyanarayana AN. V, Saha S, Keerthana HS, Naresh A, Girase YK, Chatterjee T. Synlett 2024; 35: 2465