

Subscribe to RSS
DOI: 10.1055/a-2410-2039
A Practical Guide to SuFEx Chemistry: An Overview of S(VI)-SuFEx Linkers and Their Reactivity
Authors
We thank the Universita degli Studi di Bari Aldo Moro and the Consorzio Interuniversitario Nazionale sulle Metodologie e Processi Innovativi di Sintesi (CINMPIS consortium). D.S. and R.L. acknowledge funding from the European Commission's Horizon Europe research and innovation programme through the Marie Skłodowska-Curie doctoral network "GreenDigiPharma" (grant agreement No 101073089). P.N. acknowledges funding from the European Commission's Horizon Europe research and innovation programme through a Marie Skłodowska-Curie Postdoctoral Fellowship "ExpandFlow" (grant agreement No. 101106497).

Abstract
Sulfur(VI)–fluoride exchange (SuFEx) is a second generation, metal-free click chemistry concept introduced by Sharpless in 2014. Since the introduction of the concept, a large variety of synthetic methodologies to S(VI)-SuFEx hubs, and their derivatization with oxygen-, nitrogen-, and carbon-based nucleophiles have been developed. Herein, we provide a concise and practical overview for their preparation, SuFExability, inclusive of which substituents are tolerated. The stereoselective synthesis of aza-derivatives is also discussed.
1 Introduction
2 Synthesis of SuFEx Hubs
2.1 Sulfonyl Fluorides
2.2 Fluorosulfates
2.3 Sulfamoyl Fluorides
2.4 Sulfonimidoyl Fluorides
2.5 Sulfuramidimidoyl Fluorides and Sulfurofluoridoimidates
2.6 Sulfondiimidoyl Fluorides
3 Reactivity of SuFEx Hubs
3.1 Reactivity of Sulfonyl Fluorides
3.2 Reactivity of Fluorosulfates
3.3 Reactivity of Sulfamoyl Fluorides
3.4 Reactivity of Sulfonimidoyl Fluorides
3.5 Reactivity of Sulfurofluoridoimidates
3.6 Reactivity of Sulfondiimidoyl Fluorides
4 Stereochemical Considerations in SuFEx Chemistry
5 Conclusion and Outlook
1
Introduction

Since the introduction of the concept in 2001, Click chemistry, or alternatively coined linkage chemistry, has been a reliable methodology for the rapid assembly of molecules via simple building blocks.[1] The concept is based on mimicking biosynthetic reactions in nature. Among the various representative reactions, the copper-catalyzed azide-alkyne cycloaddition,[2] has received the most attention, particularly in drug development and molecular biology. However, its frequent requirement for transition metal catalysis stands in contrast with typical requirements in biochemistry. The most recent class of linkage chemistry however, sulfur(VI)–fluoride exchange (SuFEx) overcomes this challenge as it can proceed with naturally occurring linkage partners (e.g., phenols, anilines, etc.) under transition-metal-free conditions, thus allowing the extension of this methodology to polymer chemistry,[3] high-throughput screening,[4] inverse drug discovery,[5] antibody modification,[6] [18F]-radiolabeling,[7] or the conjugation of biologically important motifs to electrophilic warheads (Figure [1]A).[8] [9] Although in principle feasible with sulfur centers at various oxidation states, sulfur(VI) centers have received most widespread attention as it allows decoration with combinations with various heteroatoms. Nonetheless, the potential of SuFEx-ligation chemistry hinges on the unique properties of the sulfur–fluorine bond. The S–F bond is, compared to its S–Cl equivalent, characterized by a high bond strength (BDE 90 kcal mol–1),[10,11] thus tolerating hydrolysis and other synthetic routine manipulations. With targeted modulation of the reaction environment, the S–F bond can be activated towards exchange. Typical modes of activation include addition of proton/silyl source, fluorophilic Lewis acids (e.g., Ca salts), or Lewis bases (typically tertiary amines).[12] [13] [14] [15] [16] [17]

Large parts of SuFEx methodology have circled around the use of sulfonyl fluorides and it is thus inherent that their preparation, as well as reactivity towards nitrogen-, oxygen-, and carbon-based nucleophiles is well-studied. Sulfonimidoyl fluorides, the mono-aza variants, and sulfondiimidoyl fluorides, the di-aza variants, are much less explored, although the imidic group provides another point of derivatization and modulation of physiochemical and chemical properties, including the enantioselective introduction of nucleophiles into a chiral sulfur center (Figure [1]B). It is thus imperative that the development of synthetic methodologies enabling the preparation of such less explored motifs, including stereoselective methods, can have a significant impact on many areas of organic synthesis beyond medicinal chemistry. Given the rapid progress and growing applications within this area,[10] , [18] [19] [20] [21] we provide a practical guide to SuFEx in three parts. First, we share an overview for the preparation of currently accessible monofluorinated sulfur(VI) hubs, and second give a summary on their derivatization. This overview also identifies current limitations and gaps in literature, necessitating further research to further drive advancements in this area. In the third part, we discuss the N-protecting group dependent reactivity of aza-isosteres of sulfonyl fluorides.
2
Synthesis of SuFEx Hubs
2.1Sulfonyl Fluorides
A myriad of strategically differing protocols is available for the synthesis of sulfonyl fluorides from a variety of sulfur sources. A selection of protocols is discussed here, yet given the large volume of protocols, the reader is also directed to leading reviews and references therein.[10] [18] [19] , [22] [23] [24] Whereas sulfuryl fluoride has been shown to react with alkyl- and (hetero)aryl Grignard reagents (Table [1], entry 1),[25] or in situ generated arynes,[26] sulfur dioxide is also used for the conversion of (hetero)aryl boronic acids using bismuth catalysts (Table [1], entry 2).[27] Sulfonyl chlorides, on the other hand, can be converted into the corresponding sulfonyl fluorides by treatment with KHF2 (Table [1], entry 3),[12] Nonetheless, the use of gaseous precursors, or the required preparation of relatively unstable sulfonyl chlorides brings about practical challenges, which are addressed by the use of solid sulfur sources. For example, N-(chlorothio)phthalimide[28] or, more frequently, 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct (DABSO) are solid, bench-stable precursors that have received interest as precursors to sulfonyl fluorides (Table [1], entries 4–8, 11, and 12).[29] In general, these two-step transformations consist of installation of the desired aryl or alkyl group to afford a sulfinate salt, followed by oxidative electrophilic fluorination with NFSI or Selectfluor. For example, Willis reported that alkyl or (hetero)aryl Grignard reagents can react with DABSO to provide sulfonyl fluorides in up to 93% yield after oxidative fluorination (Table [1], entry 4).[30] In addition, also palladium-catalyzed protocols are suitable. For example, Willis and Ball independently reported that aryl bromides (Table [1], entry 5)[30] or aryl iodides (Table [1], entry 6),[31] respectively, are effective precursors for sulfinate salts that are oxidatively fluorinated in the same pot, enabling the synthesis of complex substrates of pharmaceutical importance (e.g., celecoxib or sildenafil).
Functionalization of DABSO is, however, not limited to polar nucleophilic addition or transition-metal-catalyzed cross-coupling pathways, but also aryl radicals have been found to react with DABSO to generate sulfinate salts that can subsequently be oxidatively fluorinated. For example, arylhydrazine hydrochloride (Table [1], entry 7)[32] or arenediazonium salts (Table [1], entry 8)[33] have been shown to release the desired reactive aryl radicals upon treatment with a copper catalyst. A transition-metal-free Sandmeyer-type reaction leverages arenediazonium salts as aryl radical precursors, and Na2S2O5 as the sulfur source (Table [1], entry 9).[34]
Mechanistically different, also sulfonamides have been reported to be suitable precursors for the synthesis of alkyl- and (hetero)aryl-substituted sulfonyl fluorides. Selective activation of the amino group by pyrylium tetrafluoroborate, allows in situ preparation of a sulfonyl chloride with MgCl2, which is then converted into sulfonyl fluoride with potassium fluoride (Table [1], entry 10).[35]
More recently, photocatalysis has opened an alternative avenue for the synthesis of alkyl-substituted sulfonyl fluorides as those are generally precluded from the previously described palladium-catalyzed protocols.[36] [37] [38] [39] [40] [41] As such, carboxylic acids (Table [1], entry 11)[41,42] and Katritzky pyridinium salt (Table [1], entry 12)[43] intermediates (readily prepared from primary amines) have been identified as suitable alkyl radical precursors, to afford the desired sulfonyl fluorides under photocatalytic conditions, although the Katritzky pyridinium salts can also be activated thermally. Notably, and in contrast to previous methods, Larionov’s decarboxylative protocol[41] allowed for the inclusion of the electrophilic fluorine source within the one-pot setup rather than later addition.
Other enabling technologies have been recently utilized to overcome some challenges faced under classical batch methodologies, including the handling of gaseous reagents. For example, De Boggraeve and co-workers reported the oxidative halogenation of thiols using a two-chamber reactor to generate ex situ chlorine gas from calcium hypochlorite in the first chamber with the thiol and KF as fluoride source in the second chamber resulting in the corresponding sulfonyl fluorides (Table [1], entry 13).[44] Thiols have also been found suitable sulfur sources for the synthesis of sulfonyl fluorides upon treatment with sodium hypochlorite and KHF2,[45] or for a more expansive scope under electrochemical conditions. With regards to the latter, Noël showed that using a carbon/iron electrode system, alkane- or (hetero)arenethiols and dialkyl or di(hetero)aryl disulfides, which can also be oxidatively fluorinated with Selectfluor,[46] can be converted into the corresponding sulfonyl fluorides; potassium fluoride is used as the fluorine source (Table [1], entry 14).[47] In a related approach, sulfonyl hydrazides were used as the precursor (Table [1], entry 15).[48]
2.2
Fluorosulfates
Fluorosulfates were first reported as early as 1930[49] yet their chemical properties and utility were not extensively investigated until Sharpless and co-workers reported a range of (hetero)aryl fluorosulfates in 2014.[12] Specifically, sulfuryl fluoride can be converted into fluorosulfates in excellent yields by exchange of a S–F bond with a range of phenols under treatment with triethylamine (Table [1], entry 16). As an alternative approach, the same group reported that aryl silyl ethers are equally suitable nucleophiles for the synthesis of aryl fluorosulfates from sulfuryl fluoride (Table [1], entry 17). Given the strong affinity between silicon and fluorine, the S–F bond is activated for exchange with a range of phenols, allowing the quantity of required base to be reduced. Although efficient for most substrates at room temperature, elevated temperatures were required for more complex substrates.[12] A conceptually similar approach was reported by Moses and co-workers, who employed HMDS as a reagent with double functionality: First, as an in situ silylating agent for phenols, and second as a hydrogen fluoride scavenger. In combination with Barton’s base (BTMG, 2-tert-butyl-1,1,3,3-tetramethylguanidine), the synthesis of fluorosulfates proceeded with significantly shorter reaction times compared to the previous two approaches (15 min vs. minimum 2 h) (Table [1], entry 18).[50] Although flow technology, as an enabling technology, offers an avenue for the sustainable and safe handling of sulfuryl fluoride gas (Table [1], entry 19)[51] handling gaseous sulfur sources remains challenging from a practical aspect. To overcome this challenge, a two-chamber reactor can be utilized in which sulfuryl fluoride is generated ex situ from 1,1′-sulfonyldiimidazole, a practical solid precursor (Table [1], entry 20).[52] In the same research line, a fluorosulfuryl imidazolium salt was introduced as another shelf-stable solid precursor. The enhanced leaving group ability allowed for short reaction times with a range of phenols in up to 98% yield (Table [1], entry 21).[53] An alternative crystalline, bench-stable reagent for the synthesis of fluorosulfates was introduced by am Ende and co-workers.[54] Notably, [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF) is prepared in a single step, and efficiently reacts with phenols within ten minutes at room temperature to afford the desired fluorosulfates in 57–97% yield (Table [1], entry 22).
2.3
Sulfamoyl Fluorides
Sulfamoyl fluorides are accessible by reaction between sulfuryl fluoride and secondary amines. DMAP (4-(dimethylamino)pyridine) was shown to be an effective activating agent for the construction of the S–N bond (Table [1], entry 23). Whereas a range of functional groups are tolerated in this process (e.g., alkynes, ethers, azides etc.), poorly nucleophilic substrates (e.g., anilines or primary amines) did not afford the product.[12] A conceptually related approach leveraging flow technology minimizes the challenges related to handling sulfuryl fluoride, and enables the synthesis of sulfamoyl fluorides from anilines (Table [1], entry 24).[51] These substrates can alternatively be prepared by a difluoro-λ3-bromane-induced Hofmann rearrangement of sulfonamides.[55] Once more overcoming the challenge of utilizing gaseous sulfuryl fluoride, imidazolium salts were found suitable precursors also for sulfamoyl fluorides, allowing the scope to be extended to primary amines (Table [1], entry 25).[53] Previously mentioned bench-stable precursor [4-(acetylamino)phenyl]imidodisulfuryl difluoride (AISF) reacted efficiently with secondary amines in up to 96%, tolerating free hydroxyl groups or terminal alkynes (Table [1], entry 26).[54] Notably, in both cases activating agents, such as DMAP, were not required. Furthermore, fluorosulfuryl isocyanate was reported as a precursor. In contrast to previous examples, the S–N bond is already formed, interestingly allowing selective modification on the isocyanate moiety with secondary amines or alcohols, before leveraging the SuFEx reactivity (Table [1], entry 27).[56]
2.4
Sulfonimidoyl Fluorides
Sulfonimidoyl fluorides, aza isosters of sulfonyl fluorides, have begun to attract considerable interest in pharmaceutical application, as the isosteric substitution of oxygen with nitrogen modulates the basicity, solubility, and reactivity of these compounds.[57] For example, Sharpless and co-workers converted gaseous thionyl tetrafluoride into the desired sulfonimidoyl fluoride via an iminosulfur oxydifluoride by stepwise sulfur–fluorine exchange with primary amines, and an organometallic at low temperatures (Table [1], entry 28).[58] In general, difluorothionyl aniline derivatives showed good selectivity for monosubstitution on the sulfur center, whereas strongly electron-withdrawing substituents (e.g., p-tolylsulfonyl group) promoted over-substitution. With respect to the organometallic reagent, a wide range of organolithiums, including several lithium heteroaryls were tolerated (e.g., pyridine, N-methylindole, benzofuran, and benzothiophene). Notably, vinyllithium and alkynyllithium were not sufficiently nucleophilic to effect the desired substitution. A much earlier method by Johnson, which bypassed the requirement for a gaseous sulfur-containing precursors, consisted of an oxidative chlorination/fluorination sequence of sulfinamides (Table [1], entry 29).[59] Despite the excellent yields obtained, this methodology is also practically challenging given its long reaction durations, as well as the requirement for the somewhat capricious synthesis of unstable sulfonimidoyl chloride as an intermediate, which is subsequently converted into the corresponding fluoride with a source of nucleophilic fluorine (NaF, KF, or TBAF). Related direct approaches, which prevent the requirement for an intermediate sulfonimidoyl chloride, and enables the synthesis of highly enantioenriched sulfonimidoyl fluorides were independently reported by Bull (Table [1], entry 30)[60] and Lopchuk (Table [1], entry 31),[61] and will be discussed more in detail in Section 4. In addition to previous reports, which relied on S(VI) or S(IV) precursors, also S(II) precursors are suitable starting materials for the synthesis of sulfonimidoyl fluorides. To this extent, treatment of N-(arylsulfenyl)phthalimide precursors with potassium fluoride affords sulfinyl trifluorides, which can then be further derivatized (Table [1], entry 32).[28] The highly electrophilic intermediate can be converted into the corresponding sulfonimidoyl fluorides by substitution with a range of primary amines.
2.5
Sulfuramidimidoyl Fluorides and Sulfurofluoridoimidates
Compared to previously introduced SuFEx linkers, the synthesis (and reactivity) of sulfuramidimidoyl fluorides and sulfurofluoridoimidates is underexplored. Nonetheless, one convergent protocol for their synthesis has been reported (Table [1], entry 33 and 34).[62] Gaseous thionyl tetrafluoride is converted into iminosulfur oxydifluoride as a common intermediate. Further sulfur–fluorine exchange with secondary amines, or aryl silyl ethers provides the desired sulfuramidimidoyl fluorides and sulfurofluoridoimidates in almost quantitative yields and notably without the requirement for purification, respectively.
2.6
Sulfondiimidoyl Fluorides
Sulfondiimidoyl fluorides are comparatively uncommon compounds, hence only a few limited methodologies were available until the seminal work by Willis, disclosed in 2022 (Table [1], entry 35). This methodology involves the multistep preparation of sulfinamidines from an asymmetric sulfurdiimide, followed by oxidative fluorination to sulfondiimidoyl fluorides. In detail, the nucleophilic addition of organometallics to sulfurdiimide and the subsequent protection of the nitrogen atom afford the corresponding sulfinamidines in good yields.[63] The oxidative step is performed on the preformed sodium salt of sulfinamidines with NFSI, enabling the preparation of N-tert-octyl-N′-nosylsulfondiimidoyl fluorides in generally good yields. An elaboration of this work was recently disclosed by the same group allowing access to a variety of different N-protecting groups.[64]
|
Entry |
SuFEx Hub |
S-source |
Substituent |
Conditions |
Ref. |
|
 |
 |
|||||
|
1 |
 Sulfonyl fluoride |
SO2F2 |
alkyl, (hetero)aryl |
 THF, rt, 1 h |
[25] |
|
|
2 |
SO2 |
aryl |
 diarylbismuth tetrafluoroborate (cat.), Selectfluor, K3PO4, 4 Å MS, CHCl3/MeCN, 70 °C, 16 h |
[27] |
||
|
heteroaryl |
 diarylbismuth tosylate (cat.), NFSI, Na2CO3, CDCl3/ H2O, 60 °C, 16 h |
[27] |
||||
|
3 |
 |
alkyl, aryl |
KHF2, MeCN/H2O, rt, 2–4 h |
[12] |
||
|
4 |
DABSO |
alkyl, (hetero)aryl |
 THF, rt, 45 min then NFSI, rt, 3 h |
[30] |
||
|
5 |
DABSO |
(hetero)aryl |
 PdCl2(AmPhos)2, Et3N, i-PrOH, 75 °C, 24 h then NFSI, rt |
[30] |
||
|
6 |
DABSO |
aryl |
 Pd(OAc)2, Et3N, PAd2Bu, i-PrOH, 75 °C, 16 h then Selectfluor, MeCN, 23 °C, 2 h |
[31] |
||
|
7 |
DABSO |
(hetero)aryl |
 NFSI, pyridine, Cu2(OH)2CO3, MeCN, 40 °C, 2 h |
[32] |
||
|
8 |
DABSO |
(hetero)aryl |
 KHF2, CuCl2, 6,6′-Me2-2,2′-bipy, MeCN, rt, 12 h |
[33] |
||
|
9 |
Na2S2O5 |
(hetero)aryl |
 Selectfluor, MeOH, 70 °C, 9 h |
[34] |
||
|
10 |
 |
alkyl, aryl |
pyrylium tetrafluoroborate, MgCl2, KF, MeCN, 60 °C then H2O |
[35] |
||
|
11 |
DABSO |
alkyl |
 acridine (cat.), NFSI, CH2Cl2, LED (400 nm), rt, 12 h |
[41] |
||
|
12 |
DABSO |
alkyl |
Katritzky pyridinium salt, HE, Et3N, 2,6-lutidine, or piperidine, DMA, blue LED or 90 °C, 16 h then NFSI, rt, 4 h |
[43] |
||
|
13 |
 |
(hetero)aryl, alkyl |
Ca(OCl)2, H2SO4, 0 °C to rt then KF, MeCN, rt, 16 h |
[44] |
||
|
14 |
 or  |
alkyl, (hetero)aryl |
undivided cell (C|Fe), KF, pyridine, MeCN/HClaq, 20 mA, rt, 6–48 h |
[47] |
||
|
15 |
 |
alkyl, (hetero)aryl |
undivided cell (C|Ni), Et3N·3HF, TBAI, CH2Cl2/DMSO, 15 mA, rt, 10 h |
[48] |
||
|
16 |
 Fluorosulfate |
SO2F2 |
(hetero)aryl |
 Et3N, CH2Cl2, rt, 2–6 h |
[12] |
|
|
17 |
(hetero)aryl |
 DBU, MeCN, rt, 4–16 h |
[12] |
|||
|
18 |
(hetero)aryl |
 BTMG, HMDS, MeCN, rt, 15 min |
[50] |
|||
|
19 |
(hetero)aryl |
 Et3N or DBU, MeCN or DMF, 0.75 mL/min, 2 min |
[51] |
|||
|
20 |
 |
(hetero)aryl |
KF, TFA, rt, 18 h then Et3N, CH2Cl2, rt, 18 h |
[52] |
||
|
21 |
 |
(hetero)aryl |
 Et3N, MeCN, rt, 1 h |
[53] |
||
|
22 |
 |
(hetero)aryl |
 DBU, THF, rt, 10 min |
[54] |
||
|
23 |
 Sulfamoyl fluoride |
SO2F2 |
alkyl |
alkyl |
 DMAP, Et3N, CH2Cl2, rt, 6–18 h |
[12] |
|
24 |
SO2F2 |
alkyl, H |
alkyl, (hetero)aryl |
 Et3N or DBU, MeCN or DMF, 0.75 mL/min, 2 min |
[51] |
|
|
25 |
 |
alkyl, H |
alkyl, aryl |
 MeCN, rt, 1–2 h |
[53] |
|
|
26 |
 |
alkyl |
alkyl |
 DBU, THF, rt, 10 min |
[54] |
|
|
27 |
 |
H |
ester or amide |
MeCN, 0 °C to rt, 5 min to 12 h |
[56] |
|
|
28 |
 Sulfonimidoyl fluoride |
 |
alkyl, aryl |
(hetero)aryl |
 Et3N, MeCN, rt, 30 min then  CPME, –78 °C, 5 min |
[58] |
|
29 |
 |
alkyl, (hetero)aryl |
aryl |
Cl2 or t-BuOCl, CCl4, 0 °C, 15 min then KF, NaF or TBAF, MeCN, rt, 1–2 h |
[59] |
|
|
30 |
 |
Boc |
aryl |
Selectfluor, KOAc, EtOH, 0 °C to rt, 24 h |
[60] |
|
|
31 |
CON(i-Pr)2 |
alkyl |
NFSI, THF, 0 °C, 1 h |
[61] |
||
|
32 |
 |
alkyl, aryl |
(hetero)aryl |
trichloroisocyanuric acid, KF, TFA, MeCN, rt, 24 h then  Et3N, MeCN, rt, 18 h |
[28] |
|
|
33 |
 Sulfurofluoridoimidate |
 |
alkyl, aryl |
aryl |
 Et3N, MeCN, rt, 30 min then  DBU, MeCN, rt, 0.5–3 h |
[62] |
|
34 |
 Sulfuramidimidoyl fluoride |
 |
alkyl, aryl |
alkyl |
 Et3N, MeCN, rt, 30 min then  MeCN, rt, 30 min |
[62] |
|
35 |
 Sulfondiimidoyl fluoride |
 |
alkyl, (hetero)aryl |
carbonyl-based substituents, Nos, cyano, Si(i-Pr)3 |
NaH, 15-crown-5, 1,4-dioxane, rt, 20 min then NFSI, rt, 30 min |
[63] |
3
Reactivity of SuFEx Hubs
3.1Reactivity of Sulfonyl Fluorides
The SuFEx reactivity of sulfonyl fluorides is well-studied with several established protocols for the conversion of the S–F bond with nitrogen-, oxygen-, and carbon-based nucleophiles. For example, sulfonamides are readily prepared from alkylamines, anilines, secondary amines, and heteroarylamines when the sulfonyl fluoride was activated by Ca(NTf2)2 (Table [2], entry 1).[16] A related, and improved strategy utilizes a combination of the same Lewis acid with a nucleophilic base DABCO and expanded the amines to include ammonia (Table [2], entry 2).[17] Mechanistic investigation reveals that Ca(NTf2)2 activates the substrate, whereas DABCO offers an additional activation of amines to enhance nucleophilicity.[65]
Mechanistically different, sulfonyl fluorides can alternatively be nucleophilically activated by 1-hydroxybenzotriazole, allowing amidation of aryl- and alkylsulfonyl fluorides with a range of primary and secondary aliphatic amines, as well as anilines (Table [2], entry 3).[15] Structurally related sulfonyl azides are obtained by treatment with trimethylsilyl azide, allowing activation of the S–F bond by the silicon-based Lewis base weakening the S–F bond (Table [2], entry 4).[66] Sulfonate esters can be prepared rather straightforwardly from the corresponding alcohols. Aliphatic alcohols and phenols can be activated either by deprotonation (Table [2], entry 5),[31] or a synergistic BTMG-HMDS catalytic system that uses Barton’s hindered guanidine base BTMG as a superb SuFEx catalyst in synergy with silicon additive hexamethyldisilazane (HMDS), once again leveraging the strong affinity of silicon for fluorine weakening the S–F bond (Table [2], entry 6).[50] The conversion of sulfonyl fluorides to sulfones, on the other hand, is enabled by a variety of mechanistically differing protocols. Within the polar domain, sulfonyl fluorides can be arylated (Table [2], entry 7),[25] or trifluoromethylated (Table [2], entry 8)[67] by Grignard reagents or TMSCF3, respectively. The latter again leverages a silicon-based Lewis base as an activator for the S–F bond. Within the radical domain, photocatalysis has enabled the sulfur–fluorine exchange into various (hetero)aryl or styrene-based substituents expanding the scope of accessible sulfones in comparison to the polar domain. Whereas various aryl-based sulfones are prepared under photocatalysis (blue LED) from arylboronic acids (Table [2], entry 9),[68] the combination of a Ru(bpy)3Cl2 with DBU and styrenes, neatly affords alkene-substituted sulfones under blue light (Table [2], entry 10).[69] Mechanistically, it is suggested that DBU nucleophilically activates the sulfonyl fluoride, which can be reduced to the sulfonyl radical. This radical readily adds to styrenes affording a stabilized benzylic radical, which can be oxidized and deprotonated to afford the vinyl sulfones.
3.2
Reactivity of Fluorosulfates
The conversion of fluorosulfates into sulfates and sulfamates with alcohols and amines, respectively is reported, whereas in contrast to sulfonyl fluorides, the reaction with carbon-based nucleophiles to sulfonate esters is hitherto unreported. Conversion into sulfates is, equivalent to the reaction of sulfonyl fluorides, achievable with a synergistic BTMG-HMDS catalytic system using Barton’s hindered guanidine base BTMG as a superb SuFEx catalyst and hexamethyldisilazane as the silicon source (Table [2], entry 11).[50] Alternatively, TMS-protected phenols undergo the desired SuFEx, by silicon-mediated S–F bond activation (Table [2], entry 12).[12] Under flow conditions, protecting group-free phenols have been found to be suitable nucleophiles for the conversion into sulfates (Table [2], entry 13).[51] For the conversion of fluorosulfates into sulfamates with amines, the same protocols as for the conversion of sulfonyl fluorides into sulfonamides are applicable. Specifically, the S–F bond can be nucleophilically activated with 1-hydroxybenzotriazole (Table [2], entry 14),[15] or by combined Lewis acid/nucleophilic activation with Ca(NTf2)2/DABCO (Table [2], entry 15).[17]
3.3
Reactivity of Sulfamoyl Fluorides
Compared to the reactivity of sulfonyl fluorides and fluorosulfates, the reactivity of sulfamoyl fluorides with respect to its SuFExability is the least well studied. In fact, SuFEx on these substrates has hitherto only been studied with nitrogen-based nucleophiles to afford sulfamides. SuFEx with oxygen-based and carbon-based nucleophiles, which are suitable substrates for SuFEx on other SuFEx hubs, have not been reported for sulfamoyl fluorides to the best of our knowledge, and present a gap in the literature. For the conversion into sulfamides, the same Lewis acid/nucleophilic base protocol (Ca(NTf2)2/DABCO) is suitable (Table [2], entry 16), allowing the introduction of secondary amines, primary amines and even free ammonia or imidazole in up to 93% yield.[17] Magnesium oxide has been found to catalyze the conversion in a similar substrate scope (Table [2], entry 17).[12] A more specific subset of sulfamoyl fluorides, sulfuryl urea derivatives, have been shown to undergo SuFEx efficiently with primary amines of higher complexity, tolerating free carboxylic acids, primary alcohols, and even glycosides (Table [2], entry 18).[56]
3.4
Reactivity of Sulfonimidoyl Fluorides
Sharpless and co-workers reported, in 2018, the synthesis of sulfonimidates from sulfonimidoyl fluorides from Si-protected phenols using DBU in acetonitrile as 60 °C for 10 h, including natural compounds such as (+)-δ-tocopherol and capsaicin (Table [2], entry 19).[58] In 2020, Zuilhof showed that the silicon protecting group was not strictly required; the reaction of N-benzoylsulfonimidates with phenol and cresol using DBU in acetonitrile at room temperature formed the corresponding sulfonimidates (Table [2], entry 20). The reaction was optimized by using DBU as a catalyst and base to increase the nucleophilicity of phenol.[70]
In a similar fashion, sulfonimidamides can be prepared. This is typically achieved by base-catalyzed sulfur–fluorine exchange of amines with sulfonimidoyl fluorides, even in an enantioenriched fashion thanks to the inclusion of lithium bromide salts (Table [2], entry 21).[60] Primary and secondary amines, as well as anilines were shown to be suitable substrates, when promoted by the use of DBU or butyllithium as a base, respectively (Table [2], entry 22).[58] Sulfur–fluorine exchange of sulfonimidoyl fluorides is, however, not limited to heteroatom-containing nucleophiles, but has been extended to carbon-based nucleophiles to afford sulfoximines. For example, Johnson[59] and Sharpless[58] independently reported that a range of sulfoximines can be prepared by the reaction with organolithium reagents, although small quantities of sulfinamide, resulting from in situ reduction were obtained as byproducts in Johnson’s report (Table [2], entry 23).[59] Building on this preliminary work, Bull developed a stereospecific variant obtaining chiral sulfoximines in up to 99% ee (Table [2], entry 24).[71] The control of several variables proved fundamental for this achievement: first, the use of Grignard reagents instead of organolithiums or organozinc reagents proved crucial as the magnesium cation could trap the fluoride anion, hence preventing racemization. Second, the choice of N-protecting group is essential, as excellent enantiospecificity was only observed with NBoc or NPiv; racemization was in turn observed with smaller groups (e.g., methyl carbamate). In addition, NCbz derivatives gave reduced yields and a remarkable loss of ee (see Section 4 for more details). This is in line with the observations made by Lopchuk on their report on the enantiospecific synthesis of sulfoximines (Table [2], entry 25).[61] The synthesis of sulfoximines from sulfonimidoyl fluorides is, however, not limited to the use of Grignard or organolithium reagents. In fact, the use of organotrifluoroborates in combination with stoichiometric quantities of trimethylsilyl triflate proved suitable for the generation of alkyl-, alkenyl-, and aryl-substituted sulfoximines bearing different N-protecting groups, including propyl, ethyl, benzyl, and phenyl groups (Table [2], entry 26).[72]
3.5
Reactivity of Sulfurofluoridoimidates
Sulfurofluoridoimidates can be readily converted into the corresponding sulfurimidates in almost quantitative yield by treatment with aryl silyl ethers and 2-(tert-butylimino)-2-(diethylamino)-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) (Table [2], entry 27).[62] Similarly, secondary amines are suitable nucleophiles to afford the corresponding sulfuramidimidates (Table [2], entry 28).[62]
3.6
Reactivity of Sulfondiimidoyl Fluorides
The substitution reaction of sulfondiimidoyl fluorides with amines has been disclosed. To this end, the use of Ca(NTf)2 as an appropriate Lewis acid proved to be indispensable for the effectiveness of the transformation (Table [2], entry 29).[63] Consequently, a library of sulfondiimidamides were prepared from S-alkyl and S-aryl sulfondiimidoyl fluorides with various cyclic and acyclic secondary amines, primary amines, arylamines, tetramethylguanidine, and ammonia. The reaction is performed in tert-amyl alcohol with mild heating (60 °C). In the same fashion, fluorine exchange with C-nucleophiles enabled the synthesis of the corresponding sulfondiimines. Using predominantly aromatic and heteroaromatic organolithium reagents, the attack on the electrophilic sulfur center returns several aza-analogues of celecoxib bearing different carbonyl-based protecting groups on nitrogen (Table [2], entry 30).[64]
|
Entry |
SuFEx Hub |
Product |
Substituent |
Conditions |
Ref |
|||
 |
 |
 |
 |
|||||
|
1 |
 Sulfonyl fluoride  |
 Sulfonamide |
(hetero) aryl, alkyl |
alkyl, H |
(hetero)aryl, alkyl |
 Ca(NTf2)2, t-amyl-OH, 60 °C, 24 h |
[16] |
|
|
2 |
aryl |
alkyl, aryl, H |
(hetero)aryl, alkyl, H |
 Ca(NTf2)2, DABCO, THF, rt, 0.5–3 h |
[17] |
|||
|
3 |
alkyl, (hetero)aryl |
alkyl, H |
alkyl, aryl |
 HOBt, TMDS, DIPEA, DMSO, rt, 24 h |
[15] |
|||
|
4 |
alkyl, aryl |
azide |
TMSN3, DBU, MeCN, rt to 50 °C, 1 h |
[66] |
||||
|
5 |
 Sulfonate |
aryl |
alkyl, (hetero)aryl |
 Cs2CO3, MeCN, rt, 1 h |
[31] |
|||
|
6 |
alkyl, (hetero)aryl |
alkyl, aryl |
 BTMG, HMDS, MeCN rt or 60 °C, 5–30 min |
[50] |
||||
|
7 |
 Sulfone |
aryl |
aryl |
 THF, rt, 2 h |
[25] |
|||
|
8 |
(hetero)aryl |
CF3 |
TMSCF3, KHF2, DMSO, rt, 0.5–2 h |
[67] |
||||
|
9 |
aryl |
aryl |
 4CzIPN, Tween 20, Rb2CO3, H2O, blue LED (30 W), rt, 12 h |
[68] |
||||
|
10 |
styryl |
styryl |
styrenes, Ru(bpy)3Cl2, DBU, MeCN, blue LED, rt, 14 h |
[69] |
||||
|
11 |
 Fluorosulfate  |
 Sulfate |
(hetero)aryl |
aryl |
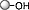 BTMG, HMDS, MeCN, rt, 30 min |
[50] |
||
|
12 |
(hetero)aryl |
aryl |
 DBU, MeCN, 4 h, rt |
[12] |
||||
|
13 |
aryl |
aryl |
 Et3N, MeCN, 0.75 mL/min, 2 min |
[51] |
||||
|
14 |
 Sulfamate |
aryl |
alkyl, H |
alkyl |
 HOBt, TMDS, DIPEA, DMSO, rt, 24 h |
[15] |
||
|
15 |
aryl |
alkyl, heteroaryl, H |
alkyl, heteroaryl |
 Ca(NTf2)2, DABCO, THF, rt, 17–24 h |
[17] |
|||
|
16 |
 Sulfamoyl fluoride  |
 Sulfamide |
alkyl |
alkyl |
alkyl, H |
alkyl |
 Ca(NTf2)2, DABCO, THF, rt, 24 h |
[17] |
|
17 |
alkyl |
alkyl |
alkyl |
alkyl |
 MgO, MeCN/H2O, reflux, 12 h |
[12] |
||
|
18 |
H |
ester or amide |
alkyl, H |
(hetero)aryl, alkyl |
 K3PO4, H2O, 80 °C, 1 M HCl |
[56] |
||
|
19 |
 Sulfonimidoyl fluoride  |
 Sulfonimidate |
alkyl, aryl |
aryl |
aryl |
 DBU, MeCN, 60 °C, 10 h |
[58] |
|
|
20 |
Bz |
aryl |
aryl |
 DBU, MeCN, rt, 0.2–24 h |
[70] |
|||
|
21 |
 Sulfonimidamide |
Boc |
aryl |
alkyl, H |
alkyl |
 Et3N, piperidine, additive (e.g., LiBr), MeCN, 80 °C, 24 h |
[60] |
|
|
22 |
aryl |
aryl |
alkyl, H |
alkyl, aryl |
 DBU, 60 °C, 24 h or |
[58] |
||
 n-BuLi, THF –78 °C, 15 min then 0 °C to rt, 1–12 h |
||||||||
|
23 |
 Sulfoximine |
alkyl, (hetero)aryl |
aryl |
alkyl, aryl |
 THF, –78 °C, 15 min |
[59] |
||
|
24 |
Boc, Piv |
aryl |
alkyl, (hetero)aryl |
 Et2O, 0 °C, 1 h |
[71] |
|||
|
25 |
CON(i-Pr)2 |
alkyl |
alkyl, (hetero)aryl |
 Et2O, –78 °C, 1 h |
[61] |
|||
|
26 |
alkyl, aryl |
alkyl, (hetero)aryl |
alkyl, (hetero)aryl |
 TMSOTf, MeCN, rt, 30 min |
[72] |
|||
|
27 |
 Sulfurofluoridoimidate  |
 Sulfurimidate |
aryl |
aryl |
aryl |
 BEMP, MeCN, rt, 1 h |
[62] |
|
|
28 |
 Sulfuramidimidate |
aryl |
aryl |
alkyl |
alkyl |
 MeCN, rt, 24 h, or DMSO, rt, 48 h |
[62] |
|
|
29 |
 Sulfondiimidoyl fluoride  |
 Sulfondiimidamide |
(hetero)aryl |
alkyl, Si(i-Pr)3, Nos, Boc, morpholinocarbonyl |
alkyl, H |
alkyl, heteroaryl, H |
 Ca(NTf2)2, t-amyl-OH, 60 °C, 24 h |
|
|
30 |
 Sulfondiimines |
aryl |
Si(i-Pr)3, Boc, morpholinocarbonyl |
alkyl, (hetero)aryl |
 THF, –78 °C or 0 °C, 1 h |
[64] |
||
a Abbreviations: BTMG = 2-tert-butyl-1,1,3,3-tetramethylguanidine; HMDS = hexamethyldisilazane; TMDS = 1,1,3,3-tetramethyldisiloxane; BEMP = 2-(tert-butylimino)-2-(diethylamino)-1,3-dimethylperhydro-1,3,2-diazaphosphorine; Nos = nosyl (4-nitrophenylsulfonyl).
4
Stereochemical Considerations in SuFEx Chemistry
The formal oxygen-to-nitrogen substitution on S(VI) SuFEx linkers necessitates focusing on reactivity and stereochemical concerns. It is worth pointing out that the nature of the group linked to the nitrogen atom of the sulfonimidoyl fluorides has a profound impact on the reactivity of such linkers. This aspect must be strictly considered when performing asymmetric synthesis. In detail, Bull recently demonstrated that it is possible to employ chiral optically active sulfonimidoyl fluorides to access sulfoximines with excellent enantiomeric excess. The reaction of N-Boc and N-Piv sulfonimidoyl fluorides with PMP-MgBr at 0 °C releases the corresponding sulfoximines in excellent yield (96%) and with complete enantioselectivity (>99% ee, Scheme [1]A).[71] Moreover, Bull reported that sulfonimidamides can also be obtained enantioselectively from N-Boc sulfonimidoyl fluorides (>99% ee). The preparation of sulfonimidoyl fluorides and sulfoximines performed with poorer selectivity when other protecting groups were employed, e.g., leading a complete loss when carbamate-substituted. Furthermore, the methodology could be applied to a selection of N-Boc-S-aryl and -S-alkyl sulfonimidoyl fluorides, but proved unsuccessful when S-tert-butyl sulfonimidoyl fluoride was used. Bull and colleagues overcame this challenge by preparing sulfoximines via sulfur alkylation and elegantly developed a stereospecific sequence to optically active sulfonimidoyl fluorides, as shown in Scheme [1]B.[71] Noteworthy, the possibility to employ sulfonimidoyl fluorides derived from S-tert-butyl sulfinamides proved strategic because: (i) both enantiomers of the sulfinamide are commercially available in high purity, (ii) the S-tert-butyl group of S-tert-butyl sulfoximines can be removed stereoselectively, releasing precious optically active S-alkyl and S-aryl sulfinamides. The recent seminal work of Lopchuk further broadened the understanding of the effect of the nitrogen protecting group over sulfonimidoyl fluoride reactivity, and efficiently employed enantiopure S-tert-butyl sulfonimidoyl fluoride for several stereospecific transformations en route to optically active aza-S(VI) compounds. Also in this case, the nature of the protecting group on the nitrogen atom proved decisive to the outcome of the reaction, and the developed methodology was broadly assessed as a reference route to enantioenriched sulfoximines and sulfonimidamides (Scheme [1]C).[61]

5
Conclusion and Outlook
In summary, we have created a concise guide summarizing currently accessible SuFEx linkers, their synthesis, and their SuFExability with different classes of nucleophiles. Although significant advances have been made towards broadening the accessibility of SuFEx-chemistry since its introduction in 2014, there remains gaps in total of four key areas which we anticipate being addressed in the foreseeable future. First, we anticipate that with increasing interest, procedures for the synthesis of sulfuramidimidoyl fluorides that avoid the requirement for gaseous thionyl tetrafluoride become available, which will also allow further in-depth studies of the reactivity of this linker. Second, we anticipate that additional SuFEx hubs will be accessible in enantiopure methods to broaden the scope. In addition, we foresee two areas for further research with respect to the reactivity of SuFEx hubs. First, the use of aliphatic alcohols as suitable nucleophiles for the transformation of SuFEx hubs has hitherto been limited by the susceptibility of the resulting product towards SN2-substitution with the fluoride ion; we expect that this limit can be overcome in the near future. Last, whereas the reactivity of heteroatom-based nucleophiles (e.g., alcohols, amines) has been well-studied for the majority of SuFEx hubs, there remains limits for carbon-based nucleophiles for some, which we expect to be addressed to broaden the scope and generality of all SuFEx-hubs for its desired application in medicinal or material chemistry.
Conflict of Interest
The authors declare no conflict of interest.
-
References
- 1 Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001; 40: 2004
- 2 Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002; 41: 2596
- 3 Xu L, Wu P, Dong JJ. New Polymers From SuFEx Click Chemistry: Syntheses and Perspectives. In RSC Polymer Chemistry Series No. 32 - Synthetic Polymer Chemistry: Innovations and Outlook. Zhao Z, Hu R, Qin A, Tang BZ. RSC; Cambridge: 2020: 1-31
- 4 Kitamura S, Zheng Q, Woehl JL, Solania A, Chen E, Dillon N, Hull MV, Kotaniguchi M, Cappiello JR, Kitamura S, Nizet V, Sharpless KB, Wolan DW. J. Am. Chem. Soc. 2020; 142: 10899
- 5 Brighty GJ, Botham RC, Li S, Nelson L, Mortenson DE, Li G, Morisseau C, Wang H, Hammock BD, Sharpless KB, Kelly JW. Nat. Chem. 2020; 12: 906
- 6 McCann HM, Lake BP. M, Hoffman KS, Davola ME, Mossman KL, Rullo AF. ACS Chem. Biol. 2022; 17: 1269
- 7 Zheng Q, Xu H, Wang H, Du W.-GH, Wang N, Xiong H, Gu Y, Noodleman L, Sharpless KB, Yang G, Wu P. J. Am. Chem. Soc. 2021; 143: 3753
- 8 Gilbert KE, Vuorinen A, Aatkar A, Pogány P, Pettinger J, Grant EK, Kirkpatrick JM, Rittinger K, House D, Burley GA, Bush JT. ACS Chem. Biol. 2023; 18: 285
- 9 Homer JA, Xu L, Kayambu N, Zheng Q, Choi EJ, Kim BM, Sharpless KB, Zuilhof H, Dong J, Moses JE. Nat. Rev. Methods Primers 2023; 3: 58
- 10 Zeng D, Deng W, Jiang X. Chem. Eur. J. 2023; 29: e202300536
- 11 Kiang T, Zare RN. J. Chem. Soc., Chem. Commun. 1980; 1228
- 12 Dong J, Krasnova L, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2014; 53: 9430
- 13 Gembus V, Marsais F, Levacher V. Synlett 2008; 1463
- 14 Gao B, Zhang L, Zheng Q, Zhou F, Klivansky LM, Lu J, Liu Y, Dong J, Wu P, Sharpless KB. Nat. Chem. 2017; 9: 1083
- 15 Wei M, Liang D, Cao X, Luo W, Ma G, Liu Z, Li L. Angew. Chem. Int. Ed. 2021; 60: 7397
- 16 Mukherjee P, Woroch CP, Cleary L, Rusznak M, Franzese RW, Reese MR, Tucker JW, Humphrey JM, Etuk SM, Kwan SC, am Ende CW, Ball ND. Org. Lett. 2018; 20: 3943
- 17 Mahapatra S, Woroch CP, Butler TW, Carneiro SN, Kwan SC, Khasnavis SR, Gu J, Dutra JK, Vetelino BC, Bellenger J, am Ende CW, Ball ND. Org. Lett. 2020; 22: 4389
- 18 Zeng D, Deng W.-P, Jiang X. Natl. Sci. Rev. 2023; 10: nwad123
- 19 Barrow AS, Smedley CJ, Zheng Q, Li S, Dong J, Moses JE. Chem. Soc. Rev. 2019; 48: 4731
- 20 Lou TS.-B, Willis MC. Nat. Rev. Chem. 2022; 6: 146
- 21 Finn MG, Kolb HC, Sharpless KB. Nat. Synth. 2022; 1: 8
- 22 Zhong T, Chen Z, Yi J, Lu G, Weng J. Chin. Chem. Lett. 2021; 32: 2736
- 23 Zheng Y, Lu W, Ma T, Huang S. Org. Chem. Front. 2024; 11: 217
- 24 Chelagha A, Louvel D, Taponard A, Berthelon R, Tlili A. Catalysts 2021; 11: 830
- 25 Lee C, Ball ND, Sammis GM. Chem. Commun. 2019; 55: 14753
- 26 Kwon J, Kim BM. Org. Lett. 2019; 21: 428
- 27 Magre M, Cornella J. J. Am. Chem. Soc. 2021; 143: 21497
- 28 Wang L, Cornella J. Angew. Chem. Int. Ed. 2020; 59: 23510
- 29 Lo PK. T, Chen Y, Willis MC. ACS Catal. 2019; 9: 10668
- 30 Davies AT, Curto JM, Bagley SW, Willis MC. Chem. Sci. 2017; 8: 1233
- 31 Tribby AL, Rodríguez I, Shariffudin S, Ball ND. J. Org. Chem. 2017; 82: 2294
- 32 Pan Q, Liu Y, Pang W, Wu J, Ma X, Hu X, Guo Y, Chen Q.-Y, Liu C. Org. Biomol. Chem. 2021; 19: 8999
- 33 Liu Y, Yu D, Guo Y, Xiao J.-C, Chen Q.-Y, Liu C. Org. Lett. 2020; 22: 2281
- 34 Zhong T, Pang M.-K, Chen Z.-D, Zhang B, Weng J, Lu G. Org. Lett. 2020; 22: 3072
- 35 Pérez-Palau M, Cornella J. Eur. J. Org. Chem. 2020; 2020: 2497
- 36 Wang P, Zhang H, Nie X, Xu T, Liao S. Nat. Commun. 2022; 13: 3370
- 37 Tilby MJ, Dewez DF, Pantaine LR. E, Hall A, Martínez-Lamenca C, Willis MC. ACS Catal. 2022; 12: 6060
- 38 Sarver PJ, Bissonnette NB, MacMillan DW. C. J. Am. Chem. Soc. 2021; 143: 9737
- 39 Chen Z.-D, Zhou X, Yi J.-T, Diao H.-J, Chen Q.-L, Lu G, Weng J. Org. Lett. 2022; 24: 2474
- 40 Zhang W, Li H, Li X, Zou Z, Huang M, Liu J, Wang X, Ni S, Pan Y, Wang Y. Nat. Commun. 2022; 13: 3515
- 41 Nguyen VT, Haug GC, Nguyen VD, Vuong NT. H, Karki GB, Arman HD, Larionov OV. Chem. Sci. 2022; 13: 4170
- 42 Ma Z, Liu Y, Ma X, Hu X, Guo Y, Chen Q.-Y, Liu C. Org. Chem. Front. 2022; 9: 1115
- 43 Andrews JA, Pantaine LR. E, Palmer CF, Poole DL, Willis MC. Org. Lett. 2021; 23: 8488
- 44 Kordnezhadian R, Zogu A, Borgarelli C, Van Lommel R, Demaerel J, De Borggraeve WM, Ismalaj E. Chem. Eur. J. 2023; 29: e202300361
- 45 Wright SW, Hallstrom KN. J. Org. Chem. 2006; 71: 1080
- 46 Kirihara M, Naito S, Ishizuka Y, Hanai H, Noguchi T. Tetrahedron Lett. 2011; 52: 3086
- 47 Laudadio G, Bartolomeu A. deA, Verwijlen LM. H. M, Cao Y, de Oliveira KT, Noël T. J. Am. Chem. Soc. 2019; 141: 11832
- 48 Park JK, Oh J, Lee S. Org. Chem. Front. 2022; 9: 3407
- 49 Lange W, Müller E. Ber. Deutsch. Chem. Ges. B 1930; 63: 2653
- 50 Smedley CJ, Homer JA, Gialelis TL, Barrow AS, Koelln RA, Moses JE. Angew. Chem. Int. Ed. 2022; 61: e202112375
- 51 Bernús M., Mazzarella D., Stanić J., Zhai Z., Yeste-Vázquez A., Boutureira O., Gargano A. F. G., Grossmann T. N., Noël T.; Nat. Synth. 2024, 3, 185.
- 52 Veryser C, Demaerel J, Bieliūnas V, Gilles P, De Borggraeve WM. Org. Lett. 2017; 19: 5244
- 53 Guo T, Meng G, Zhan X, Yang Q, Ma T, Xu L, Sharpless KB, Dong J. Angew. Chem. Int. Ed. 2018; 57: 2605
- 54 Zhou H, Mukherjee P, Liu R, Evrard E, Wang D, Humphrey JM, Butler TW, Hoth LR, Sperry JB, Sakata SK, Helal CJ, am Ende CW. Org. Lett. 2018; 20: 812
- 55 Ochiai M, Okada T, Tada N, Yoshimura A, Miyamoto K, Shiro M. J. Am. Chem. Soc. 2009; 131: 8392
- 56 Sun S, Gao B, Chen J, Sharpless KB, Dong J. Angew. Chem. Int. Ed. 2021; 60: 21195
- 57 Carneiro SN, Khasnavis SR, Lee J, Butler TW, Majmudar JD, am Ende CW, Ball ND. Org. Biomol. Chem. 2023; 21: 1356
- 58 Gao B, Li S, Wu P, Moses JE, Sharpless KB. Angew. Chem. Int. Ed. 2018; 57: 1939
- 59 Johnson CR, Bis KG, Cantillo JH, Meanwell NA, Reinhard MF. D, Zeller JR, Vonk GP. J. Org. Chem. 1983; 48: 1
- 60 Greed S, Briggs EL, Idiris FI. M, White AJ. P, Lücking U, Bull JA. Chem. Eur. J. 2020; 26: 12533
- 61 Teng S, Shultz ZP, Shan C, Wojtas L, Lopchuk JM. Nat. Chem. 2024; 16: 183
- 62 Li S, Wu P, Moses JE, Sharpless KB. Angew. Chem. Int. Ed. 2017; 56: 2903
- 63 Zhang Z.-X, Willis MC. Chem 2022; 8: 1137
- 64 Ding M, Bell C, Willis MC. Angew. Chem. Int. Ed. 2024; 63: e202409240
- 65 Han B, Khasnavis SR, Nwerem M, Bertagna M, Ball ND, Ogba OM. Inorg. Chem. 2022; 61: 9746
- 66 Barrow A, Moses J. Synlett 2016; 27: 1840
- 67 Smedley CJ, Zheng Q, Gao B, Li S, Molino A, Duivenvoorden HM, Parker BS, Wilson DJ. D, Sharpless KB, Moses JE. Angew. Chem. Int. Ed. 2019; 58: 4552
- 68 Zhen J, Li Y, Yuan H, Xu X, Du X, Li X.-Q, Luo Y. Org. Chem. Front. 2023; 10: 404
- 69 Wu X, Zhang W, Sun G, Zou X, Sang X, He Y, Gao B. Nat. Commun. 2023; 14: 5168
- 70 Liang D, Streefkerk DE, Jordaan D, Wagemakers J, Baggerman J, Zuilhof H. Angew. Chem. Int. Ed. 2020; 59: 7494
- 71 Greed S, Symes O, Bull JA. Chem. Commun. 2022; 58: 5387
- 72 Zhao S, Zeng D, Wang M, Jiang X. Nat. Commun. 2024; 15: 727
Corresponding Author
Publication History
Received: 01 August 2024
Accepted: 05 September 2024
Accepted Manuscript online:
05 September 2024
Article published online:
28 October 2024
© 2024. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001; 40: 2004
- 2 Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002; 41: 2596
- 3 Xu L, Wu P, Dong JJ. New Polymers From SuFEx Click Chemistry: Syntheses and Perspectives. In RSC Polymer Chemistry Series No. 32 - Synthetic Polymer Chemistry: Innovations and Outlook. Zhao Z, Hu R, Qin A, Tang BZ. RSC; Cambridge: 2020: 1-31
- 4 Kitamura S, Zheng Q, Woehl JL, Solania A, Chen E, Dillon N, Hull MV, Kotaniguchi M, Cappiello JR, Kitamura S, Nizet V, Sharpless KB, Wolan DW. J. Am. Chem. Soc. 2020; 142: 10899
- 5 Brighty GJ, Botham RC, Li S, Nelson L, Mortenson DE, Li G, Morisseau C, Wang H, Hammock BD, Sharpless KB, Kelly JW. Nat. Chem. 2020; 12: 906
- 6 McCann HM, Lake BP. M, Hoffman KS, Davola ME, Mossman KL, Rullo AF. ACS Chem. Biol. 2022; 17: 1269
- 7 Zheng Q, Xu H, Wang H, Du W.-GH, Wang N, Xiong H, Gu Y, Noodleman L, Sharpless KB, Yang G, Wu P. J. Am. Chem. Soc. 2021; 143: 3753
- 8 Gilbert KE, Vuorinen A, Aatkar A, Pogány P, Pettinger J, Grant EK, Kirkpatrick JM, Rittinger K, House D, Burley GA, Bush JT. ACS Chem. Biol. 2023; 18: 285
- 9 Homer JA, Xu L, Kayambu N, Zheng Q, Choi EJ, Kim BM, Sharpless KB, Zuilhof H, Dong J, Moses JE. Nat. Rev. Methods Primers 2023; 3: 58
- 10 Zeng D, Deng W, Jiang X. Chem. Eur. J. 2023; 29: e202300536
- 11 Kiang T, Zare RN. J. Chem. Soc., Chem. Commun. 1980; 1228
- 12 Dong J, Krasnova L, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2014; 53: 9430
- 13 Gembus V, Marsais F, Levacher V. Synlett 2008; 1463
- 14 Gao B, Zhang L, Zheng Q, Zhou F, Klivansky LM, Lu J, Liu Y, Dong J, Wu P, Sharpless KB. Nat. Chem. 2017; 9: 1083
- 15 Wei M, Liang D, Cao X, Luo W, Ma G, Liu Z, Li L. Angew. Chem. Int. Ed. 2021; 60: 7397
- 16 Mukherjee P, Woroch CP, Cleary L, Rusznak M, Franzese RW, Reese MR, Tucker JW, Humphrey JM, Etuk SM, Kwan SC, am Ende CW, Ball ND. Org. Lett. 2018; 20: 3943
- 17 Mahapatra S, Woroch CP, Butler TW, Carneiro SN, Kwan SC, Khasnavis SR, Gu J, Dutra JK, Vetelino BC, Bellenger J, am Ende CW, Ball ND. Org. Lett. 2020; 22: 4389
- 18 Zeng D, Deng W.-P, Jiang X. Natl. Sci. Rev. 2023; 10: nwad123
- 19 Barrow AS, Smedley CJ, Zheng Q, Li S, Dong J, Moses JE. Chem. Soc. Rev. 2019; 48: 4731
- 20 Lou TS.-B, Willis MC. Nat. Rev. Chem. 2022; 6: 146
- 21 Finn MG, Kolb HC, Sharpless KB. Nat. Synth. 2022; 1: 8
- 22 Zhong T, Chen Z, Yi J, Lu G, Weng J. Chin. Chem. Lett. 2021; 32: 2736
- 23 Zheng Y, Lu W, Ma T, Huang S. Org. Chem. Front. 2024; 11: 217
- 24 Chelagha A, Louvel D, Taponard A, Berthelon R, Tlili A. Catalysts 2021; 11: 830
- 25 Lee C, Ball ND, Sammis GM. Chem. Commun. 2019; 55: 14753
- 26 Kwon J, Kim BM. Org. Lett. 2019; 21: 428
- 27 Magre M, Cornella J. J. Am. Chem. Soc. 2021; 143: 21497
- 28 Wang L, Cornella J. Angew. Chem. Int. Ed. 2020; 59: 23510
- 29 Lo PK. T, Chen Y, Willis MC. ACS Catal. 2019; 9: 10668
- 30 Davies AT, Curto JM, Bagley SW, Willis MC. Chem. Sci. 2017; 8: 1233
- 31 Tribby AL, Rodríguez I, Shariffudin S, Ball ND. J. Org. Chem. 2017; 82: 2294
- 32 Pan Q, Liu Y, Pang W, Wu J, Ma X, Hu X, Guo Y, Chen Q.-Y, Liu C. Org. Biomol. Chem. 2021; 19: 8999
- 33 Liu Y, Yu D, Guo Y, Xiao J.-C, Chen Q.-Y, Liu C. Org. Lett. 2020; 22: 2281
- 34 Zhong T, Pang M.-K, Chen Z.-D, Zhang B, Weng J, Lu G. Org. Lett. 2020; 22: 3072
- 35 Pérez-Palau M, Cornella J. Eur. J. Org. Chem. 2020; 2020: 2497
- 36 Wang P, Zhang H, Nie X, Xu T, Liao S. Nat. Commun. 2022; 13: 3370
- 37 Tilby MJ, Dewez DF, Pantaine LR. E, Hall A, Martínez-Lamenca C, Willis MC. ACS Catal. 2022; 12: 6060
- 38 Sarver PJ, Bissonnette NB, MacMillan DW. C. J. Am. Chem. Soc. 2021; 143: 9737
- 39 Chen Z.-D, Zhou X, Yi J.-T, Diao H.-J, Chen Q.-L, Lu G, Weng J. Org. Lett. 2022; 24: 2474
- 40 Zhang W, Li H, Li X, Zou Z, Huang M, Liu J, Wang X, Ni S, Pan Y, Wang Y. Nat. Commun. 2022; 13: 3515
- 41 Nguyen VT, Haug GC, Nguyen VD, Vuong NT. H, Karki GB, Arman HD, Larionov OV. Chem. Sci. 2022; 13: 4170
- 42 Ma Z, Liu Y, Ma X, Hu X, Guo Y, Chen Q.-Y, Liu C. Org. Chem. Front. 2022; 9: 1115
- 43 Andrews JA, Pantaine LR. E, Palmer CF, Poole DL, Willis MC. Org. Lett. 2021; 23: 8488
- 44 Kordnezhadian R, Zogu A, Borgarelli C, Van Lommel R, Demaerel J, De Borggraeve WM, Ismalaj E. Chem. Eur. J. 2023; 29: e202300361
- 45 Wright SW, Hallstrom KN. J. Org. Chem. 2006; 71: 1080
- 46 Kirihara M, Naito S, Ishizuka Y, Hanai H, Noguchi T. Tetrahedron Lett. 2011; 52: 3086
- 47 Laudadio G, Bartolomeu A. deA, Verwijlen LM. H. M, Cao Y, de Oliveira KT, Noël T. J. Am. Chem. Soc. 2019; 141: 11832
- 48 Park JK, Oh J, Lee S. Org. Chem. Front. 2022; 9: 3407
- 49 Lange W, Müller E. Ber. Deutsch. Chem. Ges. B 1930; 63: 2653
- 50 Smedley CJ, Homer JA, Gialelis TL, Barrow AS, Koelln RA, Moses JE. Angew. Chem. Int. Ed. 2022; 61: e202112375
- 51 Bernús M., Mazzarella D., Stanić J., Zhai Z., Yeste-Vázquez A., Boutureira O., Gargano A. F. G., Grossmann T. N., Noël T.; Nat. Synth. 2024, 3, 185.
- 52 Veryser C, Demaerel J, Bieliūnas V, Gilles P, De Borggraeve WM. Org. Lett. 2017; 19: 5244
- 53 Guo T, Meng G, Zhan X, Yang Q, Ma T, Xu L, Sharpless KB, Dong J. Angew. Chem. Int. Ed. 2018; 57: 2605
- 54 Zhou H, Mukherjee P, Liu R, Evrard E, Wang D, Humphrey JM, Butler TW, Hoth LR, Sperry JB, Sakata SK, Helal CJ, am Ende CW. Org. Lett. 2018; 20: 812
- 55 Ochiai M, Okada T, Tada N, Yoshimura A, Miyamoto K, Shiro M. J. Am. Chem. Soc. 2009; 131: 8392
- 56 Sun S, Gao B, Chen J, Sharpless KB, Dong J. Angew. Chem. Int. Ed. 2021; 60: 21195
- 57 Carneiro SN, Khasnavis SR, Lee J, Butler TW, Majmudar JD, am Ende CW, Ball ND. Org. Biomol. Chem. 2023; 21: 1356
- 58 Gao B, Li S, Wu P, Moses JE, Sharpless KB. Angew. Chem. Int. Ed. 2018; 57: 1939
- 59 Johnson CR, Bis KG, Cantillo JH, Meanwell NA, Reinhard MF. D, Zeller JR, Vonk GP. J. Org. Chem. 1983; 48: 1
- 60 Greed S, Briggs EL, Idiris FI. M, White AJ. P, Lücking U, Bull JA. Chem. Eur. J. 2020; 26: 12533
- 61 Teng S, Shultz ZP, Shan C, Wojtas L, Lopchuk JM. Nat. Chem. 2024; 16: 183
- 62 Li S, Wu P, Moses JE, Sharpless KB. Angew. Chem. Int. Ed. 2017; 56: 2903
- 63 Zhang Z.-X, Willis MC. Chem 2022; 8: 1137
- 64 Ding M, Bell C, Willis MC. Angew. Chem. Int. Ed. 2024; 63: e202409240
- 65 Han B, Khasnavis SR, Nwerem M, Bertagna M, Ball ND, Ogba OM. Inorg. Chem. 2022; 61: 9746
- 66 Barrow A, Moses J. Synlett 2016; 27: 1840
- 67 Smedley CJ, Zheng Q, Gao B, Li S, Molino A, Duivenvoorden HM, Parker BS, Wilson DJ. D, Sharpless KB, Moses JE. Angew. Chem. Int. Ed. 2019; 58: 4552
- 68 Zhen J, Li Y, Yuan H, Xu X, Du X, Li X.-Q, Luo Y. Org. Chem. Front. 2023; 10: 404
- 69 Wu X, Zhang W, Sun G, Zou X, Sang X, He Y, Gao B. Nat. Commun. 2023; 14: 5168
- 70 Liang D, Streefkerk DE, Jordaan D, Wagemakers J, Baggerman J, Zuilhof H. Angew. Chem. Int. Ed. 2020; 59: 7494
- 71 Greed S, Symes O, Bull JA. Chem. Commun. 2022; 58: 5387
- 72 Zhao S, Zeng D, Wang M, Jiang X. Nat. Commun. 2024; 15: 727