

Subscribe to RSS
DOI: 10.1055/a-2787-1940
Ring-Restructuring of gem-Difluorocyclopropanes: Diverse Access to Four- to Nine-Membered Ring Skeletons
Authors
Funding None.


Abstract
Since their first synthesis by Atkinson in 1952, gem-difluorocyclopropanes (gem-DFCPs) have become indispensable building blocks in both organic synthesis and medicinal chemistry. These fluorinated three-membered carbocycles exhibit remarkable reactivity patterns due to their inherent ring strain and the electronic effects of fluorine substitution, which facilitate diverse ring-editing transformations. This comprehensive review systematically examines recent advancements in gem-DFCPs-mediated ring restructuring reactions, including their transformation into four-, five-, six-, and extended-membered ring systems. Particular emphasis is placed on elucidating the underlying reaction mechanisms, stereochemical control elements, and practical synthetic applications. Through this analysis, we aim to provide a structured framework that guides future research in the rapidly expanding field of fluorinated cycloaddition chemistry.
Introduction
Fluorine, with its exceptional electronegativity (4.0) and atomic radius (1.47 Å) comparable to that of hydrogen (1.2 Å), imparts unique chemical properties to fluorinated compounds.[1] [2] The introduction of fluorine atoms into organic molecules changes the structure and electronic properties of the compounds, causing profound changes in physicochemical properties. Therefore, the fluorinated ring systems, including fluorobenzenes and their derivatives, gem-difluorocyclopentanones, and gem-difluorocyclobutanes, have become ubiquitous structural motifs in natural products and indispensable pharmacophores in medicinal chemistry ([Scheme 1]).[3] [4] [5] [6] [7] For instance[8] when a fluorine atom replaces the natural hydroxyl group at the 2'-position of the deoxycytidine sugar ring of gemcitabine, a pyrimidine-based drug used for cancer treatment, the therapeutic effect will be significantly enhanced. This fluorine substitution confers resistance to degradation by cytidine deaminase, thereby substantially prolonging the drug's half-life. Moreover, the fluorine atom enables a dual antitumor mechanism: it inhibits ribonucleotide reductase, disrupting the supply of DNA synthesis precursors, while its active metabolites are incorporated into DNA strands, leading to termination of DNA elongation. Such a synergistic action markedly enhances the antitumor activity of the drug.

On the other hand, gem-DFCPs have emerged as particularly valuable synthetic intermediates due to their unique structural features: the presence of two fluorine atoms on the same carbon atom induces substantial ring strain compared with their nonfluorinated cyclopropane counterparts.[9] [10] [11] [12] [13] [14] This inherent strain not only enhances their reactivity but also facilitates selective C–C bond cleavage, allowing the construction of diverse fluorinated architectures. Consequently, the [3 + n]-cycloaddition chemistry of gem-DFCPs has attracted considerable attention in recent years, offering versatile strategies for the synthesis of complex fluorinated molecules.
The chemistry of gem-difluorocyclopropanes (gem-DFCPs) has evolved significantly since their first synthesis in 1953 by Tarrant et al, who successfully prepared gem-DFCP via the Zn-mediated reaction of 1,3-dibromo-1,1-difluoro-2-methylbutane.[15] Afterward, Taguchi et al developed the Michael-induced ring closure strategy, where activated alkenes containing BrCF2 groups were utilized in reactions with nucleophiles, and provided a unique route for the highly enantioselective synthesis of gem-DFCPs ([Fig. 1]).[16] Entering the 21st century, the emergence of new reagents greatly propelled the field forward: Zhu et al introduced TFDA (FSO2CF2CO2SiMe3) in 2000 ([Fig. 1]),[17] which was later followed by the development of MFDA (FSO2CF2CO2Me) in 2012.[18] These reagents enabled efficient transformations of electron-deficient substrates such as acrylates. In 2011, Wang et al systematically developed the Ruppert–Prakash reagent (TMSCF3/NaI) system ([Fig. 1]),[19] which has since become the most widely used method due to its excellent functional group compatibility (e.g., tolerance to Boc-amines and esters) and scalability. That same year, Li et al also reported the more reactive Me3SiCF2Br/TBAB system ([Fig. 1]).[20] More recently, to tackle the challenges posed by electron-deficient alkenes, Goswami et al pioneered a new cobalt-catalyzed TMSCF3 strategy in 2017 ([Fig. 1]).[21] Developed across different periods, these methods together constitute a versatile toolkit applicable to a broad spectrum of substrate types, strongly facilitating the exploration and application of such fluorinated structures in various cutting-edge research areas.

Over the past seven decades, these unique fluorinated cyclopropanes have become the focus of extensive synthetic and mechanistic investigations.[17] [22] [23] [24] The field has witnessed remarkable progress, evolving from early studies of homocyclization and isomerization processes[25] to contemporary breakthroughs in transition metal-catalyzed ring-opening transformations.[17] [26] A pivotal moment in the field occurred in 2003 with Dolbier and Battiste's comprehensive review, which systematically cataloged the structural features, synthetic methods, reactivity patterns, and applications of gem-DFCPs up to 2002. The field continued to advance, as evidenced by Zhu and colleagues' review (2023), which focused specifically on transition metal-catalyzed cross-coupling reactions of gem-DFCPs.[17] More recently, in 2024, Li's group contributed another significant review that highlighted innovative transformations of gem-DFCPs via double C–F bond cleavage, accompanied by in-depth analyses of design principles, selectivity control, and mechanistic insights.[26]
Despite these comprehensive reviews, there remains a notable limitation in the systematic analysis of ring-restructuring reactions of gem-DFCPs. While transition metal-catalyzed ring-opening reactions have dominated recent research efforts, the emerging field of ring-editing transformations has gained considerable momentum and warrants dedicated attention. This review, therefore, aims to address this limitation by providing a comprehensive synthesis of recent advancements in the ring-restructuring chemistry of gem-DFCPs. Through detailed mechanistic analyses and systematic classification of reaction pathways, we aim to provide a solid foundation for future innovations in this rapidly evolving field of fluorinated cyclopropane chemistry.
Synthesis of Five-Membered Rings from gem-Difluorocyclopropanes
Five-membered ring architectures represent a privileged structural motif that is widely distributed in natural products,[27] [28] [29] biologically relevant molecules,[3] [30] [31] and functional materials.[32] [33] [34] Of particular importance, functionalized five-membered carbocyclic frameworks serve as essential core units in a diverse array of biologically relevant compounds.[35] [36] [37] [38] [39] [40] [41] Recent advances in synthetic methodologies have highlighted the remarkable utility of gem-DFCPs as versatile building blocks, enabling efficient construction of these five-membered ring systems through innovative ring-opening and ring-recombination strategies.
Synthesis of gem-Difluorocyclopentanes
Zeng and Xia developed an Rh-catalyzed [3 + 2] cycloaddition reaction between gem-DFCPs and internal olefins, enabling the efficient synthesis of gem-difluorinated cyclopentanes ([Scheme 2]).[42] The reaction mechanism starts with the conversion of the Rh precursor [Rh(C2H4)2Cl]2 into the tetra-coordinated cationic Rh species 4 in the presence of BINAP, AgBF4, and an excess of olefinic substrate 2. Subsequent oxidative addition of gem-DFCPs 1 to the active Rh complex 5 generates the key gem-DFMCB intermediate 6. In the presence of excess olefinic substrate 2, the intermediate 6 coordinates to 2, and the coordinating alkene ligand migrates and inserts itself into the cross-cyclic ring from the less sterically hindered side of 7, followed by the formation of the six-membered cross-ring intermediate 8. Finally, the C(benzyl)-C(benzyl) bond undergoes reductive elimination, yielding the gem-difluorinated cyclopentane product 3 and regenerating the rhodium catalyst 5 for the subsequent catalytic cycle.

Archer et al reported a novel [3 + 2] radical cascade reaction, catalyzed by a newly discovered cationic sulfur-centered radical, promoting the transformation of 9 with olefins 10 to give gem-difluoro-2-vinylcyclopentanes 11 ([Scheme 3]).[43] The proposed reaction mechanism for this reaction involves several key steps: first, the single-electron oxidation of the thiol precursor 12 generates the reactive thiyl radical 13, which is rapidly captured by 9 to form intermediate 15 through a ring-opening/radical addition sequence. The reaction of intermediate 15 with 10 leads to the formation of intermediate 16. This is followed by a 5-exo-trig cyclization occurs, releasing the product 11 in a thermodynamically favored trans configuration and regenerating the thiyl radical 13. Finally, the catalytic cycle is completed through the reduction of the dicationic dimer 14 by IrII, regenerating both the active radical species 13 and the thiol precursor 12.

Synthesis of gem-Difluorocyclopentene
Cyclopentene frameworks are widely used in both synthetic and naturally occurring bioactive molecules. In 2014, Orr et al made a significant contribution to the field by reporting a novel thermal rearrangement of ethyl3-(2,2-difluoro-3-phenylcyclopropyl)acrylate 17 ([Scheme 4]).[44] The researchers elucidated the stereospecific nature of this transformation through meticulous 19F NMR spectroscopic analysis, and showed that the observed stereochemical results were determined by competitive stereoisomerization processes of the cyclopropane precursors prior to the vinyl cyclopropane rearrangement. Their systematic investigation revealed different mechanistic pathways for the geometric isomers: while trans-17 underwent direct rearrangement to yield 19, its cis counterpart 18 required initial isomerization to the trans configuration before proceeding to the final product.

Building upon their previous findings, the same group subsequently developed an atom-economical synthetic route to novel gem-difluorocyclopentenes 21 under practical reaction conditions (100°C), supported by advanced computational studies of the reaction mechanism ([Scheme 5]).[45] In this reaction, difluorovinylcyclopropane derivatives 20 served as key precursors, which were efficiently transformed to the target products 21 through a well-defined vinylcyclopropane rearrangement (VCPR) pathway. This methodology not only demonstrated excellent atomic efficiency but also provided a practical and scalable route to access these valuable fluorinated structures.

Zhang and Gevorgyan have reported a significant advancement in this field. They developed an innovative hydroalkenylation/diastereoselective rearrangement cascade for the construction of difluorocyclopentene scaffolds ([Scheme 6]).[46] Their systematic investigation revealed that gem-DFCPs 22 bearing phenyl and thioether substituents readily undergo hydroalkenylation with alkenes 23, followed by a facile ring-expansion cycloisomerization to afford gem-difluorocyclopentenes 24 in good yields. This methodology proved particularly effective for the synthesis of tricyclic systems, as demonstrated by the successful preparation of compounds 24c and 24e from indene derivatives. Notably, the rearrangement process exhibits remarkable diastereoselectivity, exclusively generating cis-substituted cyclopentenes, highlighting the synthetic utility of this transformation for the stereocontrolled construction of complex fluorinated architectures.

Furthermore, the domino reaction, involving the in situ generation and subsequent transformation of gem-DFCPs, was developed. Aono et al reported an efficient nickel-catalyzed reaction between silyl dienol ethers 25, derived from α,β-unsaturated ketones, and trimethylsilyl gem-difluoro-2-(fluorosulfonyl)acetate ([Scheme 7]).[47] This innovative reaction proceeds through a two-step sequence: it first enables the formation of siloxydifluorovinylcyclopropanes 27 at the electron-rich alkene moiety, which subsequently undergoes a smooth vinylcyclopropane–cyclopentene rearrangement to furnish the silyl gem-difluorocyclopent-1-en-1-yl ethers 26 in excellent yield. This cascade process not only demonstrates the synthetic versatility of gem-DFCPs but also provides a streamlined approach to accessing valuable fluorinated cyclopentene derivatives.

The same research group also developed an organocatalytic approach for the synthesis of 1-(tert-butyldimethylsilyloxy)-5,5-difluorocyclopent-1-en e 31 via the decomposition of diene 29 ([Scheme 8]).[48] This methodology employs naphthalene-1,8-diamine 32 as an organocatalyst for the generation of gem-difluorocarbene intermediates. The resulting carbene species are chemoselectively added to silyl dienol ethers to form intermediates 30, which subsequently undergo a vinylcyclopropane rearrangement to yield 31. This metal-free process shows remarkable versatility, accommodating a wide range of substrates 29 bearing either electron-donating or electron-withdrawing groups at both terminal (R1) and internal (R2) positions, and yielding the corresponding products 31a–h in excellent yields. In particular, the methodology proved effective for more complex substrates, as demonstrated by the successful transformation of a cyclohexene-containing silyl dienol ether to the bicyclic product 31g in 66% yield.

Synthesis of Cyclopent-2-en-1-ones
Cyclopent-2-en-1-ones bearing various substituents on the ring are valuable building blocks in organic synthesis. A domino reaction was developed and proved to provide an efficient access to cyclopent-2-en-1-ones via the key gem-DFCP intermediates. In 2016, Song and co-workers reported a catalytic difluorocyclopropanation of enolizable ketones 33 by using TMSCF2Br as a unique difluorocarbene source. The in situ generated siloxydifluorocyclopropanes 36 were used for the synthesis of gem-difluorocyclopentenone 35 ([Scheme 9]).[49] This process, facilitated by copper or silver, generated metal difluorohomoenolates, which then underwent intramolecular addition and elimination to efficiently yield 35.

A plausible reaction mechanism for the formation of α-ene-difluorocyclopentanone 35 can be proposed as follows: First, the reaction of 33 with Br-activated TMSCF2Br produces intermediate 36 through catalytic enolization and difluorocyclopropanation. Treatment of 36 with CuCl, assisted by a halide anion, forms copper alkoxide 38, which is then converted to copper difluorohomoenolate 39 via β-carbon elimination. The coordination of the copper in 39 then shifts from oxygen to sulfur to give the intermediate 40. This intermediate underwent addition-elimination (via 41) to afford 35 as the final product ([Scheme 9]).
In addition, Fuchibe et al discovered that silyl dienol ethers 42, derived from α,β-unsaturated ketones, underwent proton-sponge-catalyzed cyclopropanation with trimethylsilyl 2,2-difluoro-2-fluorosulfonylacetate in a regioselective manner, leading to 1,1-difluoro-2-siloxy-2-vinylcyclopropanes 43 in good yields ([Scheme 10]).[50] These cyclopropanes 43 were subsequently subjected to fluoride-ion-catalyzed ring-opening to produce 1-fluorovinyl vinyl ketones 44 (i.e., Nazarov precursors). Treatment of these precursors with Me3Si+B(OTf)4 − regioselectively promoted the Nazarov cyclization to produce the intermediate 45. The rate and regioselectivity of this cyclization were significantly enhanced by the presence of the fluoro substituent, enabling the efficient synthesis of biologically promising α-fluorocyclopentenone derivatives 46.

Synthesis of Pyrroles
Fluoropyrroles represent a crucial structural motif in numerous pharmacologically active compounds.[51] [52] [53] Yang et al developed a Brønsted acid-mediated regioselective ring-opening reaction of 47 with nitrile derivatives 48, leading to the synthesis of fluoropyrroles 49 ([Scheme 11]).[54] Their mechanistic investigation revealed that the reaction proceeds through the protonation of gem-difluorocyclopropyl ketone 50, followed by partial ring opening to generate intermediate 51. This protonation step facilitates the selective cleavage of the proximal C–C bond, as the resulting carbocation is stabilized by the adjacent fluorine atoms. Subsequent nucleophilic attack by acetonitrile and intramolecular cyclization forms the five-membered ring intermediate 52. The final product 49 is obtained through a dehydrofluorination process followed by the rearrangement of intermediate 52.

In a recent significant advancement, Wu et al reported a novel ligand-controlled regioselective ring-opening/defluorocyclization of gem-DFCPs with enaminones 53 ([Scheme 12]).[55] This palladium-catalyzed transformation demonstrates remarkable selectivity in synthesizing structurally diverse pyrroles 55 and 56 through the strategic use of different phosphine ligands. The methodology exhibits broad substrate scope, delivering various pyrrole derivatives in moderate to excellent yields.

Mechanistic studies revealed that the catalytic cycle initiates with the oxidative addition of strained gem-DFCPs to Pd(0), generating palladium complex 57. Subsequent β-F elimination produces the key allyl-Pd(II) intermediate 58. The reaction pathway then diverges based on the steric properties of the phosphine ligand: (1) when employing the sterically demanding QPhos ligand, N-coordination with enaminone 53 forms intermediate 59; (2) with the less hindered XPhos ligand, intermediate 60 is generated instead. Both pathways proceed through inner-sphere 3,3'-reductive elimination, regenerating the catalyst and yielding either branched fluoroalkene 61 or linear fluoroalkene 62. These intermediates then undergo annulation/tautomerization sequences to afford the final pyrrole products 55 and 56, respectively.
Continuing interest has been paid to the development of novel synthetic methods using gem-DFCPs. In this context, Liu et al have developed a Pd-IPent-catalyzed selective ring-opening deprotonation annulation reaction of gem-DFCPs with enamides 64 to efficiently construct multisubstituted N–H pyrrole derivatives 65 by selective cleavage of the C1–C3 bond, two C–F bonds, and the C–N bond ([Scheme 13]).[56] The reaction proceeds via oxidative addition of gem-DFCPs over a Pd(0) catalyst to form the palladium cyclic intermediate 66, which is subsequently coordinated with deprotonated enamides 64 and substance 67 to form the bis(η 1-allyl)palladium intermediate 71. Elimination by 3,3́-reduction generates the monofluorinated intermediate 72, which then undergoes intramolecular nucleophilic substitution and tautomerization to produce the N–Ac pyrrole 74. After the base-assisted deacetylation reaction, N–H pyrrole products 75 were obtained in good to excellent yields.

Synthesis of Furans
Furan derivatives represent a versatile class of heterocyclic compounds that possess diverse physicochemical properties, enabling their widespread applications across various fields, including medicinal chemistry, photochemistry, and electrochemistry.[58] [59] In a notable contribution to furan synthesis, Xu et al discovered that gem-difluorocyclopropyl aryl ketones 76 undergo a unique ring-opening/cyclization sequence in the presence of anhydrous MgI2, leading to complete defluorination and subsequent formation of furan derivatives 77 ([Scheme 14]).[60] This transformation proceeds through a sophisticated mechanism involving multiple iodide-mediated steps. First, the nucleophilic attack of the iodide ion on intermediate 78 triggers the ring-opening to generate the enol intermediate 79. Subsequent elimination of a fluorine atom yields the enone intermediate 80, followed by β-elimination of a second fluoride ion to form the allenic intermediate 81. In the absence of imine, the reaction pathway diverges through an electrophilic activation by molecular iodine (I2) of the enone intermediate 81 to give the intermediate 82. The 83 subsequently converts to intermediate 84, leading to the final iodofuran product 77 through a deprotonation process.

Moreover, Sugiishi et al demonstrated that the synthesis of 3-fluoro-2,5-disubstituted furans 85 from gem-difluorocyclopropyl ketones 84 could be efficiently achieved using trifluoro methanesulfonic acid (CF3SO3H) via a ring expansion process, facilitated by the activation of the carbonyl group in the starting material ([Scheme 15]).[61] [Scheme 15] illustrates a plausible mechanism for this transformation. Initially, CF3SO3H coordinates with the oxygen atom of the carbonyl group in compound 84, forming the activated intermediate 86. This is followed by a ring-opening reaction, which generates the benzylic carbocation intermediate 87. Subsequently, the oxygen atom of the enol then attacks the carbocation, leading to the formation of intermediate 88 through intramolecular cyclization. Finally, deprotonation and aromatization close the reaction, yielding the desired fluorofuran product 85.

Lv et al developed an efficient catalytic system for the synthesis of diverse furan derivatives 90 from readily accessible gem-DFCPs and ketones 89, utilizing sterically unencumbered Pd catalysts ([Scheme 16]).[62] The mechanistic investigation revealed a well-defined reaction pathway. The catalytic cycle was initiated by the oxidative addition of gem-DFCPs to Pd(0), accompanied by C–C bond activation and C–F bond cleavage, to generate the crucial allyl-Pd(II) complex 91. This intermediate then underwent a transmetalation reaction with enol 97, which was generated in situ through base-mediated deprotonation of ketone 89, forming the key bis(η 1-allyl) intermediate 93 along with its isomerization product 94. The sterically demanding NHC ligand facilitates the subsequent reductive elimination of 93, yielding the branched product 95 while regenerating the Pd(0) catalyst. The final furan product 90 was formed through a base-mediated sequence involving enolization, nucleophilic substitution, and rearomatization, which was promoted by the less sterically hindered Pd catalyst system. This elegant methodology, enabled by precise control of the coordination environment and optimization of reaction parameters (including base selection and reaction time), allowed for the selective synthesis of both β-monofluorinated alkenes and their corresponding furan derivatives through controlled C–F bond cleavage.

gem-DFCP diesters have emerged as novel donor–acceptor cyclopropanes, demonstrating remarkable reactivity in [3 + 2] cycloaddition reactions with a wide range of aldehydes and ketones.[63] [64] [65] [66] In this context, Liu et al pioneered the use of gem-difluoro substituents as an unconventional donor group for activating cyclopropane substrates 98 in catalytic cycloaddition reactions ([Scheme 17]).[67] This innovative approach enables the efficient assembly of various densely functionalized gem-difluorotetrahydrofuran skeletons 100 in high yields under mild reaction conditions. The proposed mechanism involves initial coordination with AlCl3 to generate intermediate 101, followed by an SN2 nucleophilic attack of aldehyde 99 on C1 of 101 to produce intermediate 102. Subsequent rotation of the C1–C3 bond and intramolecular cyclization of 102, accompanied by the dissociation of AlCl3, ultimately affords the final product 100 through the process of generation and transformation of intermediate 103.

Synthesis of Pyrazoles
Qian et al successfully developed a novel Pd/NHC-catalyzed reaction between hydrazones 104 and gem-DFCPs, enabling the synthesis of two distinct types of pyrazoles 105 and 106 through a defluorinated allylation/annulation cascade reaction ([Scheme 18]).[68] This innovative approach demonstrated remarkable regioselectivity, allowing for the specific incorporation of different carbon atoms from the gem-DFCPs into the pyrazole ring.

The proposed mechanism involves the initial oxidative addition of the Pd(0) catalyst into the gem-DFCP ring, facilitating C–C bond activation and subsequent C–F bond cleavage to generate the allyl-PdII complex 66. Subsequent N-coordination and deprotonation of the hydrazones lead to the formation of bis(η-allyl) intermediates 108 and 109. For aryl hydrazones, intermediate 108 undergoes a Pd/NHC-mediated inner-sphere 3,3'-reductive elimination, yielding the E-linear products 95. In contrast, aldehyde hydrazones (R2 = H) follow a distinct pathway, where Pd-assisted intramolecular defluorination/annulation/tautomerization affords pyrazoles 105. Interestingly, alkyl hydrazones preferentially undergo 3,3'-reductive elimination through an alternative regioisomeric pathway, producing branched products 110. Similarly, for aldehyde hydrazones (R2 = H), an analogous Pd-assisted intramolecular defluorination/annulation/tautomerization sequence leads to pyrazole product 106, with the C2 and C3 atoms of the gem-DFCPs being selectively incorporated into the pyrazole ring.
Synthesis of Six-Membered Rings from gem-difluorocyclopropanes
Six-membered rings represent a ubiquitous structural motif widely found in various bioactive natural products, pharmaceutical compounds, and functional materials.[69] [70] Recent advances in synthetic methodology have demonstrated that these crucial six-membered ring structures can be efficiently constructed through gem-DFCPs rearrangement and ring expansion reactions.
In 2020, Ahmed et al presented a novel palladium-catalyzed gem-difluorinated cyclopropane alkynylation reaction, which proceeds via C–C bond activation and C–F bond cleavage, followed by C–C coupling. This method operated under mild reaction conditions and demonstrated broad substrate compatibility, enabling both 1,1-disubstituted and complex-molecule-modified gem-difluorinated cyclopropanes to react smoothly with alkynes 114, yielding a diverse array of valuable aromatic compounds 115 ([Scheme 19]).[71] A plausible reaction mechanism, as illustrated in [Scheme 19], was proposed. The process begins with the activation of a C–C bond in the presence of a palladium catalyst and a fluorinated substrate, forming intermediate 57. Next, β-F elimination from intermediate 57 generates the π-allyl palladium complex 58, which then reacts with alkynes to produce intermediate 116. Through C–C bond elimination, monofluorinated enynes 117 are formed, releasing the palladium catalyst for further catalytic cycles. Isomerization of the skipped enyne 118 leads to the formation of the enallene intermediate 120. Finally, intramolecular C–H bond activation and 6π-electron electrocyclization afford the desired product 115.

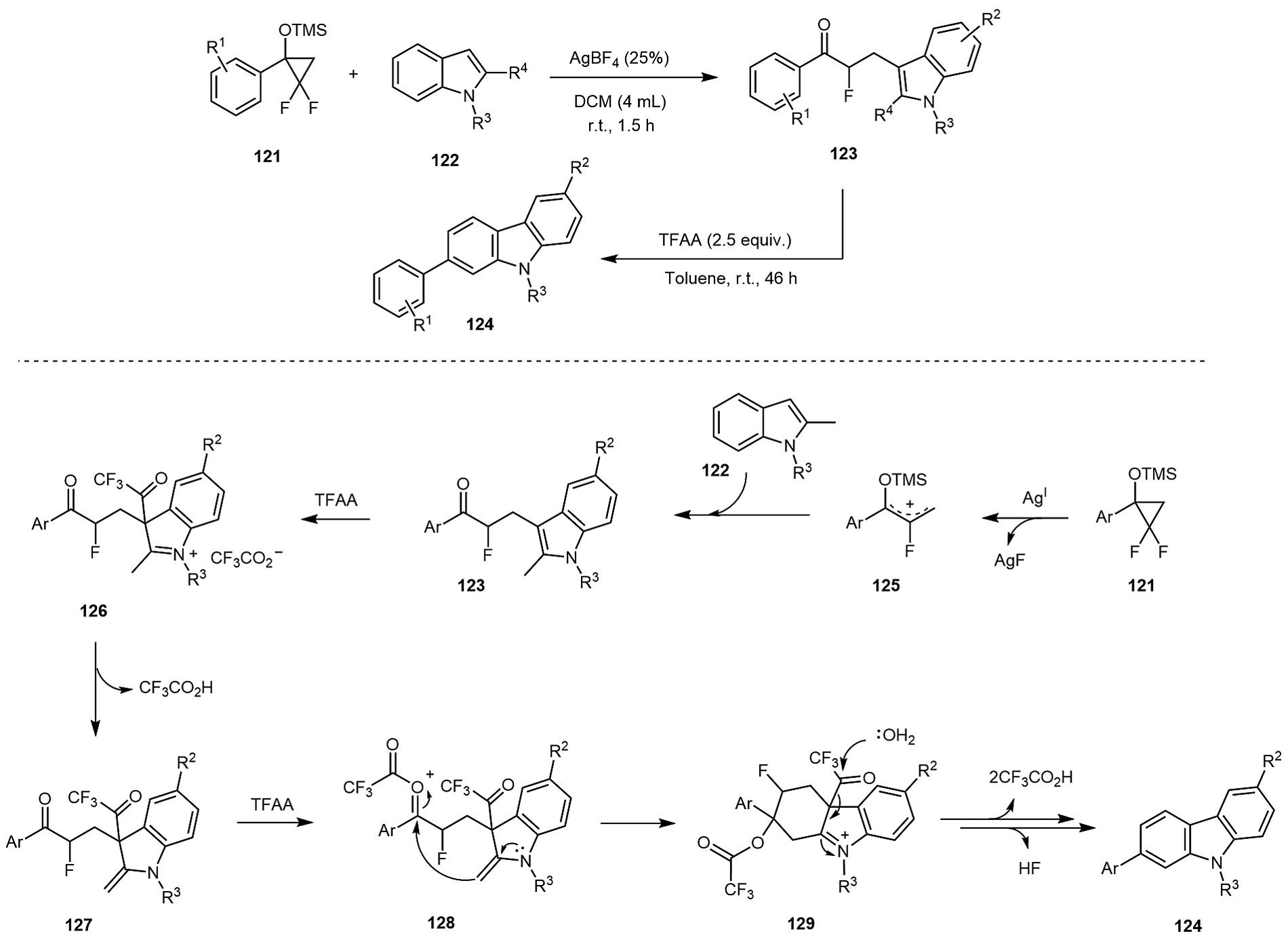
In the same year, Liu et al discovered that the defluorinated ring-opening indolylation of siloxydifluorocyclopropane 121 with indoles 122 could be catalyzed by AgBF4, yielding α-fluoro-β-indolylacetone 123. Subsequent cyclization of 123 in the presence of trifluoroacetic anhydride (TFAA) in toluene at room temperature afforded carbazoles 124 ([Scheme 20]).[72] A plausible mechanism was proposed for this transformation. The process begins with Ag(I)-mediated defluorinated ring-opening indolylation of 121 and 122, involving the formation of an allyl cation intermediate 125, followed by a Friedel–Crafts reaction to produce α-fluoro-β-indolylacetone 123. Next, acylation at the C-3 position of the indole ring in 123 by TFAA generates the 3-trifluoroacetylindole iminium intermediate 126, which isomerizes into the enamine intermediate 127. Intramolecular nucleophilic addition of the enamine onto the TFAA-activated keto carbonyl in 127 then forms intermediate 129 via 128. Finally, driven by aromatization, intermediate 129 undergoes sequential hydrolysis and elimination of both CF3CO2H and HF to yield carbazole 124 as the final product. Very recently, Fuchibe et al reported the synthesis of 2-(1,1-difluoroethyl)-2H-1,3-benzoxazines 132 through the regioselective ring-opening of aryloxy-1,1-difluorocyclopropanes 130, followed by a Friedel–Crafts-type ring-closing reaction ([Scheme 21]).[73] The reaction begins with the treatment of 130 with trifluorocarboxylic acid, leading to the regioselective cleavage of the C–C bond at the distal end of the CF2 group via protonation. This step generates the corresponding oxocarbonylium ion intermediate 131, which undergoes nucleophilic attack with nitriles, followed by Friedel − Crafts-type ring closure to afford 2-(1,1-difluoroethyl)benzoxazines 132. Under optimized conditions, this method was successfully applied to various 2-aryloxy-1,1-difluorocyclopropanes.

Other interesting work was reported by Hang et al, who explored the [3 + 2]-cycloaddition reactions of difluoro(methylidene)cyclopropanes (F2MCPs) 133 with nitronitriles 134 to access cycloaddition products 136. The resulting cycloaddition products underwent further rearrangements to yield highly substituted 3,3-difluorinated tetrahydropyridinols 135 ([Scheme 22]).[74] The proposed mechanism for this rearrangement involves a diradical process. Initially, the reaction proceeds through the formation of difluorinated spirocyclopropane isoxazolidines 136. Upon heating, the N–O bond in intermediate 136 cleaves, generating a diradical intermediate 137. This intermediate undergoes ring opening to form a new diradical species 138, which subsequently cyclizes to produce the difluorinated tetrahydropyridinol 135.

Synthesis of Other Rings from gem-difluorocyclopropanes
Medium-sized carbocycles and heterocycles have garnered significant attention in medicinal chemistry due to their diverse biological activities and pharmaceutical potential.[75] [76] Similarly, the synthesis of large-sized carbocycles and heterocycles has been achieved through innovative approaches involving gem-DFCPs rearrangement and ring expansion reactions, expanding the synthetic toolbox for complex ring systems.
Orr et al discovered that not only can the aforementioned gem-difluorovinylcyclopropane 139 undergo thermal rearrangement to form gem-difluorocyclopentene, but trans/cis-3-(2,2-difluoro-3-phenylcyclopropyl)acrylic acid ethyl ester also undergoes [3,3]-sigmatropic rearrangement at elevated temperatures (180°C). This process yields mixtures of compounds 142 and 143, which were monitored by 19F NMR spectroscopy, revealing an integration ratio of 9:1 in favor of 142 ([Scheme 23]).[44] The reaction is anticipated to involve HF elimination through rearomatization. Furthermore, heating isolated 142 at 180°C (in diphenyl ether for 17.5 hours) resulted in the formation of the more conjugated product 143, likely via a [1,5]-hydrogen shift.

Siloxydifluorocyclopropanes are recognized as highly versatile building blocks for synthesizing a wide range of difluoromethylene compounds. However, their inherent instability has posed significant challenges in the chemical exploration of fluorinated homoenolates for the preparation of α,α-difluorinated ketones. In 2015, the Amii research group addressed this issue by reporting the ring-opening reactions of silyloxydifluorocyclopropanes 145. These reactions yielded α,α-difluorocycloalkanones 146 when treated with Na2CO3 in methanol, and monofluorinated cycloalk-2-en-1-ones 147 when treated with tetrabutylammonium fluoride in THF ([Scheme 24]).[77] This approach enabled the selective generation of gem-difluorinated cycloalkanones and monofluorinated cycloalk-2-en-1-ones, providing a valuable strategy for accessing these fluorinated structures.

In the study by Kobayashi et al, the transformation of gem-difluorosubstituted bicyclo[4.1.0]heptan-1-yl acetate 147 to 2-fluorocyclohept-2-en-1-one 149 and subsequently to the 3-methoxylated analogue 152 was demonstrated to proceed through a synergistic solubilization process involving the allylic cation intermediate 153 ([Scheme 25]).[78] In contrast, nucleophilic attack by MeLi on the ester carbonyl group led to the cleavage of the cyclopropane ring, yielding ring-opening products 149 via intermediate 148. However, upon further nucleophilic attack by MeLi on the carbonyl group, only fluoroallyl alcohol 150 was isolated, with a yield of 70%.

Functionalized gem-(difluorocyclopropyl)amines exhibit significant synthetic potential. Although extensive research has been conducted on the transformations of gem-difluorocyclopropanol derivatives, studies on the reactivity of gem-(difluorocyclopropyl)amines remain limited. In 2004, Nowak et al developed a method for synthesizing the bridged gem-(difluorocyclopropyl)amines 155 from the enamines 154 and investigated their ring-opening decomposition.[79] Prolonged heating of 155 was observed to lead to their gradual disappearance, resulting in the formation of fluorinated diene amine 157 alongside the loss of a fluorine atom through the intermediate 156. Additionally, a small amount of the hydrolyzed ketone product 158 was generated as a byproduct ([Scheme 26]).

Conclusion
Over the past two decades, fluorinated cyclopropanes have emerged as a focal point of intense research interest across organic synthesis, pharmaceutical development, and materials science. Extensive investigations into gem-DFCPs have unveiled novel reactivity patterns and selective transformations, enabling efficient access to diverse fluorinated cyclic architectures. While significant progress has been made in understanding gem-DFCPs' chemistry, several key challenges and opportunities remain in the exploration of their ring-opening and ring-recombination reactions.
First, current methodologies are predominantly limited to monosubstituted gem-DFCPs, highlighting the need for innovative strategies to access sterically demanding disubstituted and multisubstituted derivatives. Second, the synthesis of other gem-difluorocarbocycles with varying ring sizes presents an exciting frontier, particularly through the incorporation of valuable unsaturated components such as alkynes, carbon monoxide, and carbenes. Finally, although transition metal-catalyzed cyclizations of gem-difluorinated carbocycles are proposed to proceed via gem-difluorinated metallocyclobutane intermediates, the precise mechanistic details of these transformations warrant further elucidation through combined experimental and computational studies.
Continued advancements in gem-DFCP chemistry are poised to make substantial contributions to the broader fields of organic synthesis and medicinal chemistry. This review aims to provide researchers with a comprehensive and accurate understanding of recent progress in this dynamic field, with the ultimate goal of inspiring innovative developments in gem-DFCPs ring-editing strategies. We anticipate that these efforts will yield transformative synthetic methodologies with significant implications for drug discovery and materials development in the near future.
Conflict of Interest
None declared.
-
References
- 1 O'Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem Soc Rev 2008; 37 (02) 308-319
- 2 Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004; 5 (05) 622-627
- 3 Hamsici S, Sardan Ekiz M, Cinar Ciftci G, Tekinay AB, Guler MO. Gemcitabine integrated nano-prodrug carrier system. Bioconjug Chem 2017; 28 (05) 1491-1498
- 4 Quintiliani M, Persoons L, Solaroli N. et al. Design, synthesis and biological evaluation of 2′-deoxy-2′,2′-difluoro-5-halouridine phosphoramidate ProTides. Bioorg Med Chem 2011; 19 (14) 4338-4345
- 5 Dhillon S. Ivosidenib: first global approval. Drugs 2018; 78 (14) 1509-1516
- 6 Zhang GF, Liu X, Zhang S, Pan B, Liu ML. Ciprofloxacin derivatives and their antibacterial activities. Eur J Med Chem 2018; 146: 599-612
- 7 Dhillon S. Melphalan flufenamide (melflufen): first approval. Drugs 2021; 81 (08) 963-969
- 8 Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol 2006; 17 (Suppl. 05) v7-v12
- 9 Henne AL, Renoll MW, Leicester HM. Aliphatic difluorides. J Am Chem Soc 1939; 61 (04) 938-940
- 10 Misani F, Speers L, Lyon AM. Synthetic studies in the field of fluorinated cyclopropanes. J Am Chem Soc 1956; 78 (12) 2801-2804
- 11 Dantzig AH, Shepard RL, Law KL. et al. Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P-450 activities. J Pharmacol Exp Ther 1999; 290 (02) 854-862
- 12 Itoh T, Kanbara M, Ohashi M. et al. gem-Difluorocyclopropane as core molecule candidate for liquid crystal compounds. J Fluor Chem 2007; 128 (10) 1112-1120
- 13 Rullière P, Cyr P, Charette AB. Difluorocarbene addition to alkenes and alkynes in continuous flow. Org Lett 2016; 18 (09) 1988-1991
- 14 Osada S, Sano S, Ueyama M, Chuman Y, Kodama H, Sakaguchi K. Fluoroalkene modification of mercaptoacetamide-based histone deacetylase inhibitors. Bioorg Med Chem 2010; 18 (02) 605-611
- 15 Tarrant P, Lovelace AM, Lilyquist MR. Free radical additions involving fluorine compounds. IV. The addition of dibromodifluoromethane to some fluoroölefins1. J Am Chem Soc 1955; 77 (10) 2783-2787
- 16 Taguchi T, Shibuya A, Sasaki H, Endo JI, Morikawa T, Shiro M. Asymmetric synthesis of difluorocyclopropanes. Tetrahedron Asymmetry 1994; 5 (08) 1423-1426
- 17 Zhu Y, Zeng Y, Jiang ZT. et al. Recent advances in transition-metal-catalyzed cross-coupling reactions of gem-difluorinated cyclopropanes. Synlett 2023; 34 (01) 1-13
- 18 Tian F, Kruger V, Bautista O. et al. A novel and highly efficient synthesis of gem-difluorocyclopropanes. Org Lett 2000; 2 (04) 563-564
- 19 Wang F, Luo T, Hu J. et al. Synthesis of gem-difluorinated cyclopropanes and cyclopropenes: trifluoromethyltrimethylsilane as a difluorocarbene source. Angew Chem Int Ed Engl 2011; 50 (31) 7153-7157
- 20 Li L, Wang F, Ni C, Hu J. Synthesis of gem-difluorocyclopropa(e)nes and O-, S-, N-, and P-difluoromethylated compounds with TMSCF2Br+ . Angew Chem Int Ed Engl 2013; 52 (47) 12390-12394
- 21 Goswami M, de Bruin B, Dzik WI. Difluorocarbene transfer from a cobalt complex to an electron-deficient alkene. Chem Commun (Camb) 2017; 53 (31) 4382-4385
- 22 Lv L, Qian H, Li Z. Catalytic diversification of gem-difluorocyclopropanes: recent advances and challenges. ChemCatChem 2022; 14 (24) e202200890
- 23 Harmata AS, Roldan BJ, Stephenson CRJ. Formal cycloadditions driven by the homolytic opening of strained, saturated ring systems. Angew Chem Int Ed Engl 2023; 62 (04) e202213003
- 24 Zeng Y, Jiang ZT, Xia Y. Selectivity in Rh-catalysis with gem-difluorinated cyclopropanes. Chem Commun (Camb) 2024; 60 (28) 3764-3773
- 25 Dolbier Jr WR, Battiste MA. Structure, synthesis, and chemical reactions of fluorinated cyclopropanes and cyclopropenes. Chem Rev 2003; 103 (04) 1071-1098
- 26 Lv L, Su J, Li Z. Recent developments in the ring-opening transformations of gem-difluorocyclopropanes. Org Chem Front 2024; 11: 6518-6533
- 27 Hetrick KJ, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural product discovery in the genomic era. Curr Opin Chem Biol 2017; 38: 36-44
- 28 Ge Y, Czekster CM, Miller OK, Botting CH, Schwarz-Linek U, Naismith JH. Insights into the mechanism of the cyanobactin heterocyclase enzyme. Biochemistry 2019; 58 (16) 2125-2132
- 29 van Pée KH, Ligon JM. Biosynthesis of pyrrolnitrin and other phenylpyrrole derivatives by bacteria. Nat Prod Rep 2000; 17 (02) 157-164
- 30 Keam SJ. Pirtobrutinib: first approval. Drugs 2023; 83 (06) 547-553
- 31 Duggan S, Al-Salama ZT. Leniolisib: first approval. Drugs 2023; 83 (10) 943-948
- 32 Borges A, Solomon GC. Effects of aromaticity and connectivity on the conductance of five-membered rings. J Phys Chem 2017; 121 (15) 8272-8279
- 33 Jacobse PH, Daugherty MC, Čerņevičs KN. et al. Five-membered rings create off-zero modes in nanographene. ACS Nano 2023; 17 (24) 24901-24909
- 34 Di Giovannantonio M, Urgel JI, Beser U. et al. On-surface synthesis of indenofluorene polymers by oxidative five-membered ring formation. J Am Chem Soc 2018; 140 (10) 3532-3536
- 35 Cho JH, Coats SJ, Schinazi RF. Synthesis of carbocyclic nucleoside analogs with five-membered heterocyclic nucleobases. Tetrahedron Lett 2015; 56 (23) 3587-3590
- 36 Ojeda-Porras AC, Roy V, Agrofoglio LA. Chemical approaches to carbocyclic nucleosides. Chem Rec 2022; 22 (05) e202100307
- 37 Koba Y, Hirata Y, Ueda A. et al. Synthesis of chiral five-membered carbocyclic ring amino acids with an acetal moiety and helical conformations of its homo-chiral homopeptides. Biopolymers 2016; 106 (04) 555-562
- 38 Balaban AT, Oniciu DC, Katritzky AR. Aromaticity as a cornerstone of heterocyclic chemistry. Chem Rev 2004; 104 (05) 2777-2812
- 39 Gulevich AV, Dudnik AS, Chernyak N, Gevorgyan V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem Rev 2013; 113 (05) 3084-3213
- 40 Kaur N. Synthesis of six-membered N-heterocycles using ruthenium catalysts. Catal Lett 2019; 149: 1513-1559
- 41 Kaur N. Solid-phase synthesis of sulfur containing heterocycles. J Sulfur Chem 2018; 39 (05) 544-577
- 42 Zeng Y, Xia Y. Rhodium-catalyzed regio- and diastereoselective [3+2] cycloaddition of gem-difluorinated cyclopropanes with internal olefins. Angew Chem Int Ed Engl 2023; 62 (32) e202307129
- 43 Archer G, Cavalère P, Médebielle M, Merad J. Photoredox generation of isothiouronyl radical cations: a new platform in covalent radical catalysis. Angew Chem Int Ed Engl 2022; 61 (32) e202205596
- 44 Orr D, Percy JM, Tuttle T, Kennedy AR, Harrison ZA. Evaluating the thermal vinylcyclopropane rearrangement (VCPR) as a practical method for the synthesis of difluorinated cyclopentenes: experimental and computational studies of rearrangement stereospecificity. Chemistry 2014; 20 (44) 14305-14316
- 45 Orr D, Percy JM, Harrison ZA. A computational triage approach to the synthesis of novel difluorocyclopentenes and fluorinated cycloheptadienes using thermal rearrangements. Chem Sci (Camb) 2016; 7 (10) 6369-6380
- 46 Zhang Z, Gevorgyan V. Palladium hydride-enabled hydroalkenylation of strained molecules. J Am Chem Soc 2022; 144 (45) 20875-20883
- 47 Aono T, Sasagawa H, Fuchibe K, Ichikawa J. Regioselective synthesis of α,α-difluorocyclopentanone derivatives: domino nickel-catalyzed difluorocyclopropanation/ring-expansion sequence of silyl dienol ethers. Org Lett 2015; 17 (23) 5736-5739
- 48 Takayama R, Fuchibe K, Ichikawa J. Metal-free synthesis of α,α-difluorocyclopentanone derivatives via regioselective difluorocyclopropanation/VCP rearrangement of silyl dienol ethers. Akivoc 2017; 2018 (02) 72-80
- 49 Song X, Tian S, Zhao Z, Zhu D, Wang M. Controlled ring-opening of siloxydifluorocyclopropanes for carbocyclization: synthesis of difluorocyclopentenones. Org Lett 2016; 18 (14) 3414-3417
- 50 Fuchibe K, Takayama R, Yokoyama T, Ichikawa J. Regioselective synthesis of α-fluorinated cyclopentenones by organocatalytic difluorocyclopropanation and fluorine-directed and fluorine-activated Nazarov cyclization. Chemistry 2017; 23 (12) 2831-2838
- 51 Bovy PR, Reitz DB, Collins JT. et al. Nonpeptide angiotensin II antagonists: N-phenyl-1H-pyrrole derivatives are angiotensin II receptor antagonists. J Med Chem 1993; 36 (01) 101-110
- 52 Wilkerson WW, Galbraith W, Gans-Brangs K. et al. Antiinflammatory 4,5-diarylpyrroles: synthesis and QSAR. J Med Chem 1994; 37 (07) 988-998
- 53 Ruebsam F, Webber SE, Tran MT. et al. Pyrrolo[1,2-b]pyridazin-2-ones as potent inhibitors of HCV NS5B polymerase. Bioorg Med Chem Lett 2008; 18 (12) 3616-3621
- 54 Yang TP, Lin JH, Chen QY, Xiao JC. A novel reaction of gem-difluorocyclopropyl ketones with nitriles leading to 2-fluoropyrroles. Chem Commun (Camb) 2013; 49 (84) 9833-9835
- 55 Wu TS, Hao YJ, Cai ZJ, Ji SJ. Ligand-controlled regioselective cascade C–C/C–F cleavage/annulation of gem-DFCPs: a divergent synthesis of pyrroles. Org Chem Front 2024; 11 (04) 1057-1061
- 56 Liu W, Ma Y, Huang Q, Sheng J, Lv L, Li Z. Pd-IPent-catalyzed defluorinative annulation of gem-difluorocyclopropanes with enamides: synthesis of multisubstituted N-H pyrroles. Org Lett 2025; 27 (09) 2151-2156
- 57 Teponno RB, Kusari S, Spiteller M. Recent advances in research on lignans and neolignans. Nat Prod Rep 2016; 33 (09) 1044-1092
- 58 Pan JY, Chen SL, Yang MH, Wu J, Sinkkonen J, Zou K. An update on lignans: natural products and synthesis. Nat Prod Rep 2009; 26 (10) 1251-1292
- 59 Saleem M, Kim HJ, Ali MS, Lee YS. An update on bioactive plant lignans. Nat Prod Rep 2005; 22 (06) 696-716
- 60 Xu W, Ghiviriga I, Chen QY, Dolbier WR. Magnesium iodide promoted defluorinative reactions of 2, 2-difluorocyclopropyl aryl ketones with aryl imines: a new, general synthesis of 2-alkylideneazetidines. J Fluor Chem 2010; 131 (09) 958-963
- 61 Sugiishi T, Matsumura C, Amii H. Synthesis of 3-fluoro-2,5-disubstituted furans through ring expansion of gem-difluorocyclopropyl ketones. Org Biomol Chem 2020; 18 (18) 3459-3462
- 62 Lv L, Qian H, Ma Y, Huang S, Yan X, Li Z. Ligand-controlled regioselective and chemodivergent defluorinative functionalization of gem-difluorocyclopropanes with simple ketones. Chem Sci (Camb) 2021; 12 (47) 15511-15518
- 63 Pohlhaus PD, Johnson JS. Enantiospecific Sn(II)- and Sn(IV)-catalyzed cycloadditions of aldehydes and donor-acceptor cyclopropanes. J Am Chem Soc 2005; 127 (46) 16014-16015
- 64 Parsons AT, Johnson JS. Catalytic enantioselective synthesis of tetrahydrofurans: a dynamic kinetic asymmetric [3 + 2] cycloaddition of racemic cyclopropanes and aldehydes. J Am Chem Soc 2009; 131 (09) 3122-3123
- 65 Yang P, Shen Y, Feng M, Yang GS, Chai Z. Lewis acid catalyzed [3+2] annulation of γ-butyrolactone fused cyclopropane with aldehydes/ketones. Eur J Org Chem 2018; 30: 4103-4112
- 66 Pohlhaus PD, Johnson JS. Highly diastereoselective synthesis of tetrahydrofurans via Lewis acid-catalyzed cyclopropane/aldehyde cycloadditions. J Org Chem 2005; 70 (03) 1057-1059
- 67 Liu H, Tian L, Wang H. et al. A novel type of donor-acceptor cyclopropane with fluorine as the donor: (3 + 2)-cycloadditions with carbonyls. Chem Sci (Camb) 2022; 13 (09) 2686-2691
- 68 Qian H, Nguyen HD, Lv L, Chen S, Li Z. Chemo-, stereo- and regioselective fluoroallylation/annulation of hydrazones with gem-difluorocyclopropanes via tunable palladium/NHC catalysis. Angew Chem Int Ed Engl 2023; 62 (23) e202303271
- 69 Li Y, Shi H, Yin G. Synthetic techniques for thermodynamically disfavoured substituted six-membered rings. Nat Rev Chem 2024; 8 (07) 535-550
- 70 Pasternak ARO, Bechthold A, Zechel DL. Identification of genes essential for sulfamate and fluorine incorporation during nucleocidin biosynthesis. ChemBioChem 2022; 23 (15) e202200140
- 71 Ahmed EMA, Suliman AMY, Gong TJ, Fu Y. Access to divergent fluorinated enynes and arenes via palladium-catalyzed ring-opening alkynylation of gem-difluorinated cyclopropanes. Org Lett 2020; 22 (04) 1414-1419
- 72 Liu XW, Du DX, Li ST, Wang X, Xu C, Wang M. Defluorinative ring-opening indolylation of siloxydifluorocyclopropanes: controlled synthesis of α-fluoro-β-indolylpropanones for carbazole construction. Adv Synth Catal 2020; 362 (22) 5135-5140
- 73 Fuchibe K, Matsuo T, Ichikawa J. Synthesis of 2-difluoroethylated 2H-1,3-benzoxazines via proton-mediated ring opening/interrupted Ritter reaction of 1,1-difluorocyclopropanes. Org Lett 2023; 25 (23) 4276-4280
- 74 Hang XC, Chen QY, Xiao JC. 1,3-Dipolar cycloaddition of difluoro(methylene)cyclopropanes with nitrones: efficient synthesis of 3,3-difluorinated tetrahydropyridinols. Synlett 2008; 13: 989-1992
- 75 Noor R, Zahoor AF, Naqvi SAR, Haq A, Akhtar R. Synthetic potential of ring expansions of 5-membered carbo-& heterocycles: a review. Synth Commun 2022; 52: 949-973
- 76 Noor R, Zahoor AF, Mansha A. et al. Synthetic potential of regio-and stereoselective ring expansion reactions of six-membered carbo-and heterocyclic ring systems: a review. Int J Mol Sci 2023; 24 (07) 6692
- 77 Kageshima Y, Suzuki C, Oshiro K, Amii H. Highly controlled ring-opening of siloxydifluorocyclopropanes: a versatile route to cyclic fluoroketones. Synlett 2015; 26 (01) 63-66
- 78 Kobayashi Y, Taguchi T, Mamada M, Shimizu H, Murohashi J. Studies on organic fluorine compounds. XXX. Ring opening reaction of acetoxydifluorocyclopropanes with various nucleophiles. Chem Pharm Bull (Tokyo) 1979; 27 (12) 3123-3129
- 79 Nowak I, Cannon JF, Robins MJ. Synthesis and properties of gem-(difluorocyclopropyl)amine derivatives of bicyclo[n.1.0]alkanes. Org Lett 2004; 6 (25) 4767-4770
Address for correspondence
Publication History
Received: 31 March 2025
Accepted: 13 January 2026
Article published online:
16 February 2026
© 2026. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 O'Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem Soc Rev 2008; 37 (02) 308-319
- 2 Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004; 5 (05) 622-627
- 3 Hamsici S, Sardan Ekiz M, Cinar Ciftci G, Tekinay AB, Guler MO. Gemcitabine integrated nano-prodrug carrier system. Bioconjug Chem 2017; 28 (05) 1491-1498
- 4 Quintiliani M, Persoons L, Solaroli N. et al. Design, synthesis and biological evaluation of 2′-deoxy-2′,2′-difluoro-5-halouridine phosphoramidate ProTides. Bioorg Med Chem 2011; 19 (14) 4338-4345
- 5 Dhillon S. Ivosidenib: first global approval. Drugs 2018; 78 (14) 1509-1516
- 6 Zhang GF, Liu X, Zhang S, Pan B, Liu ML. Ciprofloxacin derivatives and their antibacterial activities. Eur J Med Chem 2018; 146: 599-612
- 7 Dhillon S. Melphalan flufenamide (melflufen): first approval. Drugs 2021; 81 (08) 963-969
- 8 Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol 2006; 17 (Suppl. 05) v7-v12
- 9 Henne AL, Renoll MW, Leicester HM. Aliphatic difluorides. J Am Chem Soc 1939; 61 (04) 938-940
- 10 Misani F, Speers L, Lyon AM. Synthetic studies in the field of fluorinated cyclopropanes. J Am Chem Soc 1956; 78 (12) 2801-2804
- 11 Dantzig AH, Shepard RL, Law KL. et al. Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P-450 activities. J Pharmacol Exp Ther 1999; 290 (02) 854-862
- 12 Itoh T, Kanbara M, Ohashi M. et al. gem-Difluorocyclopropane as core molecule candidate for liquid crystal compounds. J Fluor Chem 2007; 128 (10) 1112-1120
- 13 Rullière P, Cyr P, Charette AB. Difluorocarbene addition to alkenes and alkynes in continuous flow. Org Lett 2016; 18 (09) 1988-1991
- 14 Osada S, Sano S, Ueyama M, Chuman Y, Kodama H, Sakaguchi K. Fluoroalkene modification of mercaptoacetamide-based histone deacetylase inhibitors. Bioorg Med Chem 2010; 18 (02) 605-611
- 15 Tarrant P, Lovelace AM, Lilyquist MR. Free radical additions involving fluorine compounds. IV. The addition of dibromodifluoromethane to some fluoroölefins1. J Am Chem Soc 1955; 77 (10) 2783-2787
- 16 Taguchi T, Shibuya A, Sasaki H, Endo JI, Morikawa T, Shiro M. Asymmetric synthesis of difluorocyclopropanes. Tetrahedron Asymmetry 1994; 5 (08) 1423-1426
- 17 Zhu Y, Zeng Y, Jiang ZT. et al. Recent advances in transition-metal-catalyzed cross-coupling reactions of gem-difluorinated cyclopropanes. Synlett 2023; 34 (01) 1-13
- 18 Tian F, Kruger V, Bautista O. et al. A novel and highly efficient synthesis of gem-difluorocyclopropanes. Org Lett 2000; 2 (04) 563-564
- 19 Wang F, Luo T, Hu J. et al. Synthesis of gem-difluorinated cyclopropanes and cyclopropenes: trifluoromethyltrimethylsilane as a difluorocarbene source. Angew Chem Int Ed Engl 2011; 50 (31) 7153-7157
- 20 Li L, Wang F, Ni C, Hu J. Synthesis of gem-difluorocyclopropa(e)nes and O-, S-, N-, and P-difluoromethylated compounds with TMSCF2Br+ . Angew Chem Int Ed Engl 2013; 52 (47) 12390-12394
- 21 Goswami M, de Bruin B, Dzik WI. Difluorocarbene transfer from a cobalt complex to an electron-deficient alkene. Chem Commun (Camb) 2017; 53 (31) 4382-4385
- 22 Lv L, Qian H, Li Z. Catalytic diversification of gem-difluorocyclopropanes: recent advances and challenges. ChemCatChem 2022; 14 (24) e202200890
- 23 Harmata AS, Roldan BJ, Stephenson CRJ. Formal cycloadditions driven by the homolytic opening of strained, saturated ring systems. Angew Chem Int Ed Engl 2023; 62 (04) e202213003
- 24 Zeng Y, Jiang ZT, Xia Y. Selectivity in Rh-catalysis with gem-difluorinated cyclopropanes. Chem Commun (Camb) 2024; 60 (28) 3764-3773
- 25 Dolbier Jr WR, Battiste MA. Structure, synthesis, and chemical reactions of fluorinated cyclopropanes and cyclopropenes. Chem Rev 2003; 103 (04) 1071-1098
- 26 Lv L, Su J, Li Z. Recent developments in the ring-opening transformations of gem-difluorocyclopropanes. Org Chem Front 2024; 11: 6518-6533
- 27 Hetrick KJ, van der Donk WA. Ribosomally synthesized and post-translationally modified peptide natural product discovery in the genomic era. Curr Opin Chem Biol 2017; 38: 36-44
- 28 Ge Y, Czekster CM, Miller OK, Botting CH, Schwarz-Linek U, Naismith JH. Insights into the mechanism of the cyanobactin heterocyclase enzyme. Biochemistry 2019; 58 (16) 2125-2132
- 29 van Pée KH, Ligon JM. Biosynthesis of pyrrolnitrin and other phenylpyrrole derivatives by bacteria. Nat Prod Rep 2000; 17 (02) 157-164
- 30 Keam SJ. Pirtobrutinib: first approval. Drugs 2023; 83 (06) 547-553
- 31 Duggan S, Al-Salama ZT. Leniolisib: first approval. Drugs 2023; 83 (10) 943-948
- 32 Borges A, Solomon GC. Effects of aromaticity and connectivity on the conductance of five-membered rings. J Phys Chem 2017; 121 (15) 8272-8279
- 33 Jacobse PH, Daugherty MC, Čerņevičs KN. et al. Five-membered rings create off-zero modes in nanographene. ACS Nano 2023; 17 (24) 24901-24909
- 34 Di Giovannantonio M, Urgel JI, Beser U. et al. On-surface synthesis of indenofluorene polymers by oxidative five-membered ring formation. J Am Chem Soc 2018; 140 (10) 3532-3536
- 35 Cho JH, Coats SJ, Schinazi RF. Synthesis of carbocyclic nucleoside analogs with five-membered heterocyclic nucleobases. Tetrahedron Lett 2015; 56 (23) 3587-3590
- 36 Ojeda-Porras AC, Roy V, Agrofoglio LA. Chemical approaches to carbocyclic nucleosides. Chem Rec 2022; 22 (05) e202100307
- 37 Koba Y, Hirata Y, Ueda A. et al. Synthesis of chiral five-membered carbocyclic ring amino acids with an acetal moiety and helical conformations of its homo-chiral homopeptides. Biopolymers 2016; 106 (04) 555-562
- 38 Balaban AT, Oniciu DC, Katritzky AR. Aromaticity as a cornerstone of heterocyclic chemistry. Chem Rev 2004; 104 (05) 2777-2812
- 39 Gulevich AV, Dudnik AS, Chernyak N, Gevorgyan V. Transition metal-mediated synthesis of monocyclic aromatic heterocycles. Chem Rev 2013; 113 (05) 3084-3213
- 40 Kaur N. Synthesis of six-membered N-heterocycles using ruthenium catalysts. Catal Lett 2019; 149: 1513-1559
- 41 Kaur N. Solid-phase synthesis of sulfur containing heterocycles. J Sulfur Chem 2018; 39 (05) 544-577
- 42 Zeng Y, Xia Y. Rhodium-catalyzed regio- and diastereoselective [3+2] cycloaddition of gem-difluorinated cyclopropanes with internal olefins. Angew Chem Int Ed Engl 2023; 62 (32) e202307129
- 43 Archer G, Cavalère P, Médebielle M, Merad J. Photoredox generation of isothiouronyl radical cations: a new platform in covalent radical catalysis. Angew Chem Int Ed Engl 2022; 61 (32) e202205596
- 44 Orr D, Percy JM, Tuttle T, Kennedy AR, Harrison ZA. Evaluating the thermal vinylcyclopropane rearrangement (VCPR) as a practical method for the synthesis of difluorinated cyclopentenes: experimental and computational studies of rearrangement stereospecificity. Chemistry 2014; 20 (44) 14305-14316
- 45 Orr D, Percy JM, Harrison ZA. A computational triage approach to the synthesis of novel difluorocyclopentenes and fluorinated cycloheptadienes using thermal rearrangements. Chem Sci (Camb) 2016; 7 (10) 6369-6380
- 46 Zhang Z, Gevorgyan V. Palladium hydride-enabled hydroalkenylation of strained molecules. J Am Chem Soc 2022; 144 (45) 20875-20883
- 47 Aono T, Sasagawa H, Fuchibe K, Ichikawa J. Regioselective synthesis of α,α-difluorocyclopentanone derivatives: domino nickel-catalyzed difluorocyclopropanation/ring-expansion sequence of silyl dienol ethers. Org Lett 2015; 17 (23) 5736-5739
- 48 Takayama R, Fuchibe K, Ichikawa J. Metal-free synthesis of α,α-difluorocyclopentanone derivatives via regioselective difluorocyclopropanation/VCP rearrangement of silyl dienol ethers. Akivoc 2017; 2018 (02) 72-80
- 49 Song X, Tian S, Zhao Z, Zhu D, Wang M. Controlled ring-opening of siloxydifluorocyclopropanes for carbocyclization: synthesis of difluorocyclopentenones. Org Lett 2016; 18 (14) 3414-3417
- 50 Fuchibe K, Takayama R, Yokoyama T, Ichikawa J. Regioselective synthesis of α-fluorinated cyclopentenones by organocatalytic difluorocyclopropanation and fluorine-directed and fluorine-activated Nazarov cyclization. Chemistry 2017; 23 (12) 2831-2838
- 51 Bovy PR, Reitz DB, Collins JT. et al. Nonpeptide angiotensin II antagonists: N-phenyl-1H-pyrrole derivatives are angiotensin II receptor antagonists. J Med Chem 1993; 36 (01) 101-110
- 52 Wilkerson WW, Galbraith W, Gans-Brangs K. et al. Antiinflammatory 4,5-diarylpyrroles: synthesis and QSAR. J Med Chem 1994; 37 (07) 988-998
- 53 Ruebsam F, Webber SE, Tran MT. et al. Pyrrolo[1,2-b]pyridazin-2-ones as potent inhibitors of HCV NS5B polymerase. Bioorg Med Chem Lett 2008; 18 (12) 3616-3621
- 54 Yang TP, Lin JH, Chen QY, Xiao JC. A novel reaction of gem-difluorocyclopropyl ketones with nitriles leading to 2-fluoropyrroles. Chem Commun (Camb) 2013; 49 (84) 9833-9835
- 55 Wu TS, Hao YJ, Cai ZJ, Ji SJ. Ligand-controlled regioselective cascade C–C/C–F cleavage/annulation of gem-DFCPs: a divergent synthesis of pyrroles. Org Chem Front 2024; 11 (04) 1057-1061
- 56 Liu W, Ma Y, Huang Q, Sheng J, Lv L, Li Z. Pd-IPent-catalyzed defluorinative annulation of gem-difluorocyclopropanes with enamides: synthesis of multisubstituted N-H pyrroles. Org Lett 2025; 27 (09) 2151-2156
- 57 Teponno RB, Kusari S, Spiteller M. Recent advances in research on lignans and neolignans. Nat Prod Rep 2016; 33 (09) 1044-1092
- 58 Pan JY, Chen SL, Yang MH, Wu J, Sinkkonen J, Zou K. An update on lignans: natural products and synthesis. Nat Prod Rep 2009; 26 (10) 1251-1292
- 59 Saleem M, Kim HJ, Ali MS, Lee YS. An update on bioactive plant lignans. Nat Prod Rep 2005; 22 (06) 696-716
- 60 Xu W, Ghiviriga I, Chen QY, Dolbier WR. Magnesium iodide promoted defluorinative reactions of 2, 2-difluorocyclopropyl aryl ketones with aryl imines: a new, general synthesis of 2-alkylideneazetidines. J Fluor Chem 2010; 131 (09) 958-963
- 61 Sugiishi T, Matsumura C, Amii H. Synthesis of 3-fluoro-2,5-disubstituted furans through ring expansion of gem-difluorocyclopropyl ketones. Org Biomol Chem 2020; 18 (18) 3459-3462
- 62 Lv L, Qian H, Ma Y, Huang S, Yan X, Li Z. Ligand-controlled regioselective and chemodivergent defluorinative functionalization of gem-difluorocyclopropanes with simple ketones. Chem Sci (Camb) 2021; 12 (47) 15511-15518
- 63 Pohlhaus PD, Johnson JS. Enantiospecific Sn(II)- and Sn(IV)-catalyzed cycloadditions of aldehydes and donor-acceptor cyclopropanes. J Am Chem Soc 2005; 127 (46) 16014-16015
- 64 Parsons AT, Johnson JS. Catalytic enantioselective synthesis of tetrahydrofurans: a dynamic kinetic asymmetric [3 + 2] cycloaddition of racemic cyclopropanes and aldehydes. J Am Chem Soc 2009; 131 (09) 3122-3123
- 65 Yang P, Shen Y, Feng M, Yang GS, Chai Z. Lewis acid catalyzed [3+2] annulation of γ-butyrolactone fused cyclopropane with aldehydes/ketones. Eur J Org Chem 2018; 30: 4103-4112
- 66 Pohlhaus PD, Johnson JS. Highly diastereoselective synthesis of tetrahydrofurans via Lewis acid-catalyzed cyclopropane/aldehyde cycloadditions. J Org Chem 2005; 70 (03) 1057-1059
- 67 Liu H, Tian L, Wang H. et al. A novel type of donor-acceptor cyclopropane with fluorine as the donor: (3 + 2)-cycloadditions with carbonyls. Chem Sci (Camb) 2022; 13 (09) 2686-2691
- 68 Qian H, Nguyen HD, Lv L, Chen S, Li Z. Chemo-, stereo- and regioselective fluoroallylation/annulation of hydrazones with gem-difluorocyclopropanes via tunable palladium/NHC catalysis. Angew Chem Int Ed Engl 2023; 62 (23) e202303271
- 69 Li Y, Shi H, Yin G. Synthetic techniques for thermodynamically disfavoured substituted six-membered rings. Nat Rev Chem 2024; 8 (07) 535-550
- 70 Pasternak ARO, Bechthold A, Zechel DL. Identification of genes essential for sulfamate and fluorine incorporation during nucleocidin biosynthesis. ChemBioChem 2022; 23 (15) e202200140
- 71 Ahmed EMA, Suliman AMY, Gong TJ, Fu Y. Access to divergent fluorinated enynes and arenes via palladium-catalyzed ring-opening alkynylation of gem-difluorinated cyclopropanes. Org Lett 2020; 22 (04) 1414-1419
- 72 Liu XW, Du DX, Li ST, Wang X, Xu C, Wang M. Defluorinative ring-opening indolylation of siloxydifluorocyclopropanes: controlled synthesis of α-fluoro-β-indolylpropanones for carbazole construction. Adv Synth Catal 2020; 362 (22) 5135-5140
- 73 Fuchibe K, Matsuo T, Ichikawa J. Synthesis of 2-difluoroethylated 2H-1,3-benzoxazines via proton-mediated ring opening/interrupted Ritter reaction of 1,1-difluorocyclopropanes. Org Lett 2023; 25 (23) 4276-4280
- 74 Hang XC, Chen QY, Xiao JC. 1,3-Dipolar cycloaddition of difluoro(methylene)cyclopropanes with nitrones: efficient synthesis of 3,3-difluorinated tetrahydropyridinols. Synlett 2008; 13: 989-1992
- 75 Noor R, Zahoor AF, Naqvi SAR, Haq A, Akhtar R. Synthetic potential of ring expansions of 5-membered carbo-& heterocycles: a review. Synth Commun 2022; 52: 949-973
- 76 Noor R, Zahoor AF, Mansha A. et al. Synthetic potential of regio-and stereoselective ring expansion reactions of six-membered carbo-and heterocyclic ring systems: a review. Int J Mol Sci 2023; 24 (07) 6692
- 77 Kageshima Y, Suzuki C, Oshiro K, Amii H. Highly controlled ring-opening of siloxydifluorocyclopropanes: a versatile route to cyclic fluoroketones. Synlett 2015; 26 (01) 63-66
- 78 Kobayashi Y, Taguchi T, Mamada M, Shimizu H, Murohashi J. Studies on organic fluorine compounds. XXX. Ring opening reaction of acetoxydifluorocyclopropanes with various nucleophiles. Chem Pharm Bull (Tokyo) 1979; 27 (12) 3123-3129
- 79 Nowak I, Cannon JF, Robins MJ. Synthesis and properties of gem-(difluorocyclopropyl)amine derivatives of bicyclo[n.1.0]alkanes. Org Lett 2004; 6 (25) 4767-4770