

Subscribe to RSS
DOI: 10.1055/s-0043-1764218
Application of Chiral Piperidine Scaffolds in Drug Design
Authors
Funding We gratefully acknowledge financial supports from the National Science and Technology Major Project (Grant No. 2018ZX09711002-002-009), the National Natural Science Foundation of China (Grant No. 81703358), the Science and Technology Commission of Shanghai Municipality (Grant No. 17431903900, 18QB1404200, 21S11908000, 22ZR1460300), and the Graduate Innovation Fund Project of China State Institute of Pharmaceutical Industry (Grant No. YJS2021013, YJS2021011).


Abstract
Chiral piperidine scaffolds are prevalent as the common cores of a large number of active pharmaceuticals in medical chemistry. This review outlined the diversity of chiral piperidine scaffolds in recently approved drugs, and also covers the scaffolds in leads and drug candidates. The significance of chiral piperidine scaffolds in drug design is also discussed in this article. With the introduction of chiral piperidine scaffolds into small molecules, the exploration of drug-like molecules can be benefitted from the following aspect: (1) modulating the physicochemical properties; (2) enhancing the biological activities and selectivity; (3) improving pharmacokinetic properties; and (4) reducing the cardiac hERG toxicity. Given above, chiral piperidine-based discovery of small molecules will be a promising strategy to enrich our molecules' library to fight against diseases.
Keywords
chiral piperidine scaffolds - drug-like - drug molecules - drug design - medicinal chemistryIntroduction
Chiral drugs has attracted more and more attention due to their perfect adaptability to protein-binding sites. The chiral state of a molecule can be achieved by introducing a chiral center to the structure, and owing to this fact, their druggability may be greatly influenced.[1] Piperidine, as a six-membered nitrogenous heterocyclic ring, is widely present in the structure of approved drugs.[2] Thus, the study of stereochemistry of piperidine scaffolds has attracted a great deal of interest since the late last century, and has become a hot topic recently.[3] According to the U.S. Food and Drug Administration data, there are 245 drugs approved from 2015 to June 2020, including small molecules and macromolecules. Among them, the number of chiral piperidine-containing drugs is nine, including avycaz (1), cotellic (2), varubi (3), zejula (4), daurismo (5), galafold (6), akynzeo (7), ubrelvy (8), and recarbrio (9) ([Table 1], [Fig. 1]).[4] Although the introduction of chiral centers in piperidine scaffolds is often associated with increased number of synthetic steps and unusual reaction procedures, and ultimately leads to an increase in synthesis effort,[5] [6] medical researchers believe that the increased expenditure is worth because of the expected favorable effects on physicochemical properties, potency, selectivity, and pharmacokinetic (PK) profile that may be induced by the chiral piperidine scaffolds. Thus, this review aimed to provide a broad perspective on the latest advances of chiral piperidine scaffolds in medicinal chemistry. The study would assist drug discoverers to rationally design molecules for various diseases.

Abbreviations: AML, acute myeloid leukemia; UTI, urinary tract infection.
Application of Chiral Piperidine Scaffolds in Drug Design
Modulating the Physicochemical Properties
Physicochemical properties of a compound can be predicted by the parameters of pK a, logD, and logP, and methods for improving the physicochemical properties of drugs include introducing hydrophilic or lipophilic groups, changing charge state and their spatial configurations to form intermolecular or intramolecular forces, etc.[7] [8] The piperidine ring originally is a kind of groups with properties of both hydrophilicity and lipophilicity. The introduction of chiral centers in the piperidine ring, changing the position of a substituent, or introducing another substituent may alter its physicochemical properties effectively.
In 2012, Ndungu et al reported a series of measles virus RNA-dependent RNA polymerase (MeV-RdRp) inhibitors.[8] In their earlier work, they identified 10 as a potent and selective MeV-RdRp inhibitor, which exhibited an excellent activity (EC50 = 14 nmol/L) but suffered from poor water solubility and low oral bioavailability. Further structure–activity relationship (SAR) studies showed that introducing a substituent at the 2-position of the piperidine ring could effectively enhance the aqueous solubility of this series of compounds. Then, optimization of in vivo potency and aqueous solubility of compound 10 led to the discovery of compound 11, which showed an improved aqueous solubility of 60 μg/mL and a maintained MeV-RdRp inhibition (EC50 = 60 nmol/L) ([Fig. 2]).

SUCNR1 (succinate receptor 1, initially named GPR91) is a G-protein-coupled receptor, and identifies succinate as its endogenous ligand.[9] Succinate is responsible for ATP formation and energy supply,[10] and is normally located in the mitochondria, but under certain pathological conditions, it acts as a signaling molecule, and is recognized as a danger signal by SUCNR1.[10] [11] In the process of exploring a new class of SUCNR1 inhibitors, Velcicky et al discovered that both 4-piperidyl analog (12) and 3-piperidyl analog (13) had a good SUCNR1 inhibition.[12] However, the logD (pH 7.4) values of the two compounds varied a lot ([Table 2]), and compound 13 showed quite remarkable increase in the permeability and lipophilicity when compared with 12. Further research revealed that the two compounds populated remarkably different shapes in an aqueous solution ([Fig. 3]).[13] Compound 12 preferred an elongated conformation, while compound 13 was folded with an intramolecular salt bridge formed between the piperidine and the carboxylic acid moieties.[12]

|
|
|||
|---|---|---|---|
|
Compound |
R |
hSUCNR1 GTPγS (μmol/L) |
logD7.4 |
|
12 |
|
0.14 |
2.5 |
|
13 |
|
0.52 |
3.6 |



Influence on the Biological Activity
Improving the Biological Activity
Chiral centers are widely found in approved drugs and drug candidates, and have shown unique privileges in enhancing drug efficacy. Due to the asymmetry of organisms, the biological properties of chiral molecules are more abundant than achiral molecules.[14] Currently, the study of stereochemistry of the piperidine scaffold has fascinated chemists because of its profound effect on biological activity of a drug.[15] As is shown in a wide range of research studies, introducing a chiral center in the piperidine ring can not only provide researchers with more chiral piperidine-containing compounds, which is essential to improve the activity of the drugs, but also make the compound have more configurational isomers to fit in the cavum of protein.[15]
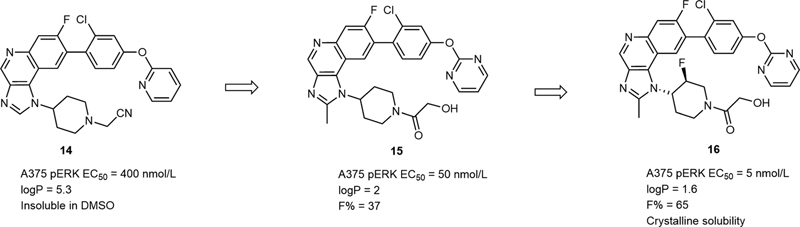
The mitogen-activated protein kinase (MAPK) signaling pathway has four branches. Among which, the extracellular regulated protein kinases (ERK) pathway plays an important role in the development of tumors.[16] In this pathway, there are three important target proteins, including Ras, Raf, and mitogen-activated protein (MEK). Many research studies suggested that inhibition of MEK may suppress the activation of ERK effectively, which is essential to the blockage of this pathway.[16] [17] In the process of identifying a new series of MEK1/2 inhibitors, the imidazoquinoline core was considered, and Poddutoori et al discovered 1-(piperidin-4-yl)-1H-imidazo[4,5-c]quinoline (14) as a high-throughput screening hit, which had an acceptable MEK1/2 inhibition (EC50 = 400 nmol/L).[18] The structure-based design led to the discovery of analogue 15, whose MEK1/2 inhibition is eightfold higher than 14. Further optimization focused on the modification of the piperidine ring of 15, with a fluorine introduced at the 3-position of the piperidine ring, to give chiral 16, which exhibited a further improved potency (EC50 = 5 nmol/L) and aqueous solubility (logP = 1.6) as well as high oral bioavailability (F% = 65) ([Fig. 4]). The crystal structure of MEK1 in complex with 16 revealed that the 3-substitued piperidine side chain can fit into the cavum of MEK easily, which is essential to increase the drug potency ([Fig. 5]).

In exploration of a new series of CVF-Bb (Cobra Venom Factor binds factor B) inhibitors, Mainolfi et al discovered the significance of R groups on the potency of CVF-Bb inhibition of a compound ([Table 3]).[19] When the R group is a morpholine ring or a piperidine ring with no chiral center, the potency was low. Taking compound 18 as an example, it had an IC50 value of 50 μmol/L; however, when a phenyl was introduced at 2-position of the piperidine ring of 18, compound 19 was obtained, with a great increase in CVF-Bb inhibitory potency (IC50 = 5.9 μmol/L). Further optimization led to the discovery of 20 and 21, whose potencies are approximately 170-fold higher than 19. It is noteworthy that the introduction of an ethoxy group at the 4-position of the piperidine ring provided 21 with approximately twofold potency higher than 20, which also proved the good effect of introducing a chiral center in the piperidine ring. Then, the replacement of methyl with a methoxy led to 22, whose potency was further increased and PK properties are good ([Fig. 6]).

|
|
|||
|---|---|---|---|
|
Compound |
R |
CVF-Bb IC50 (μmol/L) |
|
|
17 |
|
>100 |
|
|
18 |
|
50 |
|
|
19 |
|
5.9 |
|
|
20 |
|
0.033 |
|
|
21 |
|
0.018 |
|





P53 is a kind of tumor suppressor protein that maintains the integrity of the genome in a cell.[20] Human double minute 2 (HDM2) is a ubiquitin protein ligase that negatively regulates p53 and lessens its transcriptional activity as well as promotes p53 protein degradation.[20] [21] Disrupting the HDM2-p53 protein–protein interaction (PPI) with a small molecule has been recognized as a potential way in cancer therapies.[22] In 2014, Ma et al reported the discovery of a new series of HDM2-p53 PPI inhibitors.[21] Structure-based design led to the discovery of 23 that had a piperidine-containing core and an acceptable potency (IC50 = 169 nmol/L). Further optimization of 23 led to compound 24, which was a potent HDM2-p53 PPI inhibitor (IC50 = 41 nmol/L) with an allyl at the 2-position of its piperidine ring. Compared with 23, compound 24 showed an improved inhibition toward HDM2-p53 PPI and a favorable toxicity window between wt-p53 and mutant-p53 cell lines ([Fig. 7]).

In 2017, Rutaganira et al reported the discovery of a new series of calcium-dependent protein kinase 1 (CDPK1) inhibitors.[23] Among which, compound 27 showed the best potency with an IC50 value of 10.9 nmol/L (compared with 25 and 26, whose IC50 values are 14.15 and 77.5 nmol/L), suggesting an important role of two fluorine atoms at 3-position of the piperidine ring ([Table 4]). Interestingly, 27 produced a chiral center at the 4-position of the piperidine ring. The skeleton of 27 fits well into the cavum ([Fig. 8a]), observed by the crystal structure of CDPK1 in complex with 27 (PDB code: 5W9E), and the introduction of a chiral center made the nitrogen of the piperidine ring form two salt bridges with the GLU135 and GLU178 residues of CDPK1 ([Fig. 8b]).

|
|
||
|---|---|---|
|
Compound |
R |
CDPK1 IC50 (nmol/L) |
|
25 |
|
14.15 |
|
26 |
|
77.5 |
|
27 |
|
10.9 |




Glucagon-like peptide-1 receptor (GLP-1R) is the most important incretin in type-2 diabetes therapy. GLP-1R enhances the release of glucose-dependent insulin and suppresses the apoptosis of islet β cells.[24] However, the half-life time of this native peptide in blood is very low (1 − 2 minutes) as it can be mainly rapidly degraded by circulating dipeptidyl peptidase (DPP-4).[25] Therefore, it is of great importance to develop a potent GLP-1R agonist. In 2021, Decara et al discovered a new series of GLP-1R agonists that contain a 1,2,4-oxadiazole core and a substituted piperidine ring.[25] Among which, the position of the substituent on the piperidine ring mattered much to their activity. When a substituent is present at 4-position of the piperidine ring (28), a poor GLP-1 potentiation (E max = 24%) was achieved. When the substituent is morpholine-1-methyl and at 3-position of the piperidine ring (31), the GLP-1 potentiation of the compound was increased to 60% ([Table 5]).




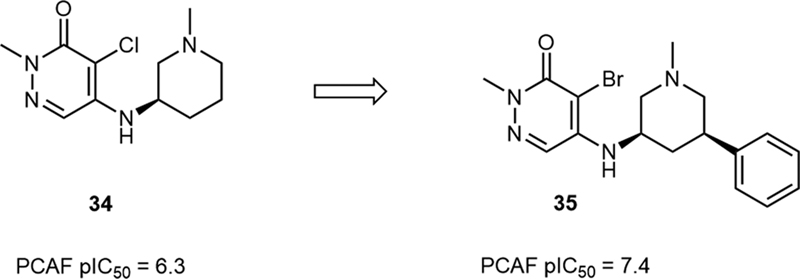
In 2016, Humphreys et al reported the discovery of a series of p300/CBP and p300/CBP associated factor (PCAF) inhibitors that had a pyridazin-3(2H)-one core, among which, the type of R groups was of great significance to their activities.[26] When the R group was a pyrrolidine (32) or a 4-substituted piperidine (33), the potencies of compounds were low. But when the substitution site in the piperidine ring changed from 4 to 3 (34), the potencies of compounds improved a lot ([Table 6]). Further SAR research studies discovered that introducing a phenyl at 5-position of the piperidine ring can further increase its activity, which led to 35, a potent PCAF inhibitor with a pIC50 value of 7.4 and exceptional selectivity over the bromodomain and extra-terminal domain (BET) bromodomain family ([Fig. 9]).
|
|
||
|---|---|---|
|
Compound |
R |
PCAF pIC50 |
|
32 |
|
4.8 |
|
33 |
|
4.8 |
|
34 |
|
6.3 |




In 2019, Huang et al discovered a series of PCAF inhibitors that had a similar structure to that of Humphreys et al.[27] The results also proved that the introduction of a chiral center in the piperidine ring improves the potencies of the series of compounds ([Table 7]). As shown in [Table 7], when the R group was a symmetric piperidine-containing structure (36, 37, and 38), the K D values of the compounds were above 200 nmol/L. But when the substituent was at the 3- and 5-position of the piperidine ring (39), the K D value increased to approximately 150 nmol/L. Further optimization led to 40, a potent PCAF inhibitor that had similar structure to 39 and a further increased potency ([Fig. 10]).

|
|
||
|---|---|---|
|
Compound |
R |
K D (μmol/L) |
|
36 |
|
0.215 |
|
37 |
|
2.16 |
|
38 |
|
1.19 |
|
39 |
|
0.152 |





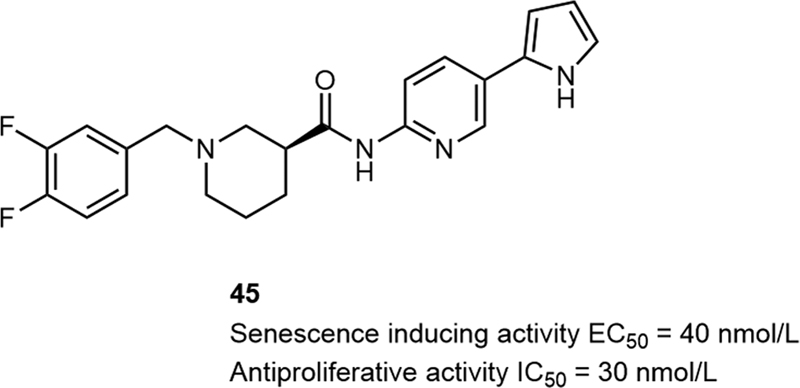
Cellular senescence exists widely in living beings, which has recently been considered as a potent strategy to suppress cancer progression.[28] [29] In 2020, Oh et al reported the discovery of a new class of senescence-inducing small molecules with antimelanoma activities in vitro.[30] High-throughput screening and high-content screening led to the identification of compound 44, a senescence inducer with good antimelanoma activities in vitro. SAR research studies suggested a great significance of the 1,3-substituent piperidine moiety in improving the potency of the compound. When the 1,3-disubstituent on the piperidine moiety was replaced by an azetidine (41), pyrrolidine (42), or symmetric piperidine (43) moiety, the activities of the compounds decreased a lot ([Table 8]). Further modification focused on the replacement of the thiazole moiety, which led to the identification of 45 ([Fig. 11]), a potent cellular senescence inducer with remarkable antimelanoma activity in vitro (EC50 = 40 nmol/L, IC50 = 30 nmol/L).
|
|
|||
|---|---|---|---|
|
Compound |
R |
Activity (μmol/L) |
|
|
EC50 |
IC50 |
||
|
41 |
|
> 20 |
> 20 |
|
42 |
|
8.00 |
10.0 |
|
43 |
|
19.0 |
> 20 |
|
44 |
|
1.24 |
0.88 |





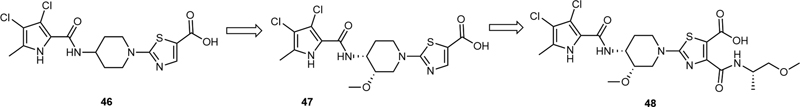
In 2014, Basarab et al reported a series of pyrrolamide topoisomerase II inhibitors.[31] Among them, compound 46 had a (piperidin-1-yl)thiazole-5-carboxylic acid moiety, and displayed DNA gyrase inhibitory potency and antibacterial activity toward gram-positive pathogens. Compound 46 functions by inhibiting the type II bacterial topoisomerases, DNA gyrase, and topoisomerase IV (Topo IV). To further improve its activity against topoisomerase II of bacteria, a methoxy group was introduced at 3-position of the piperidine ring to obtain 47, which showed an improved inhibition to a wide range of gram-positive and gram-negative bacteria ([Table 9]). Further modification was focused on the 4-position of the thiazole ring. For example, the introduction of (S)-((1-methoxypropan-2-yl)carbamoyl)thiazole-5-carboxylic acid unit led to 48, which was proved to exhibit a further improved activity against bacterial growth.
Abbreviations: Eco, E. coli; Hin, H. influenzae; Mca, M. catarrhalis; MRQR Sau, methicillin resistant, quinolone resistant S. aureus; MSSA, methicillin sensitive S. aureus; ND, not determined; Spn, S. pneumonia; Spy, S. pyogenes.
Regulating the Target Selectivity of Drug Molecules
In 2018, Karlsson et al identified a series of new fibrinolysis inhibitors.[32] They found that introducing a chiral center in the piperidine ring of the series of compounds can effectively increase their selectivity over GABAa ([Table 10]). Structure-based virtual screen led to the piperidinyl-substituted isoxazol-3-ol 49, a fibrinolysis inhibitor with high potency but low selectivity over GABAa. Further research led to 50 that had a methyl at the 2-position of the piperidine ring. As is shown in [Table 10], the potency of 50 decreased but the selectivity over GABAa remarkably increased. Then, the methyl group at 2-position of the piperidine ring was replaced by a neopentyl to give 51, which had a further increased potency, yet maintained selectivity over GABAa.

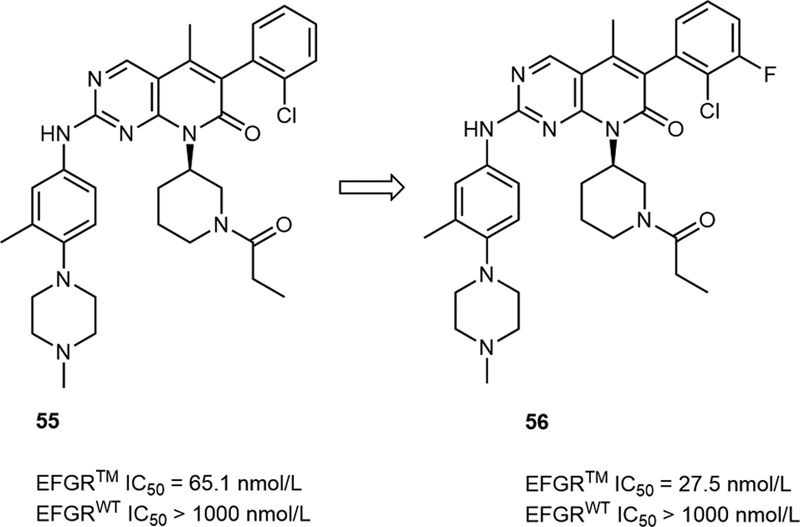
In 2019, Shen et al reported the identification of a new series of EGFRL858R/T790M/C797S inhibitors.[33] They discovered that the type of R groups was of great significance to their selectivity over EGFRWT. When the R group is a cyclohexanamine-containing moiety (52 and 53) or a symmetrical piperidine moiety (54), the selectivity of this series of compounds was low. But when the site of substitution was changed from 4-position to 3-position, their selectivity over EGFRWT increased remarkably (55, [Table 11]). At last, a fluorine substituent was introduced at 3-position of the phenyl and its potency was further increased (56, [Fig. 12]).




Improving PK Properties of Drug Molecules
Absorption, distribution, metabolism, and excretion properties are key indexes evaluating the druggability of drugs. Piperidine rings, especially chiral piperidine rings, are usually introduced to improve PK properties (e.g., half-life, clearance, bioavailability) of a drug, and thus a common structural unit existing in drugs.[34]
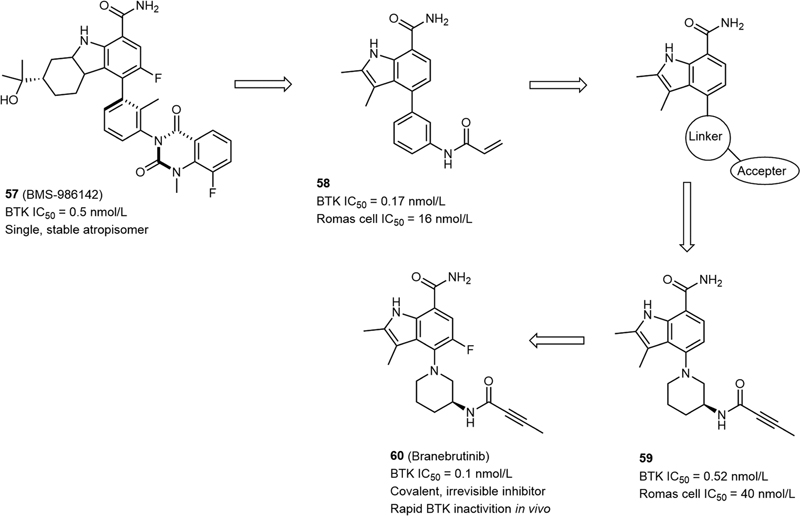
Bruton's tyrosine kinase (BTK), a member of the Tec family of nonreceptor cytoplasmic tyrosine kinases, plays an essential role in B cell receptor signaling pathways and Fcγ receptor signaling in leukocytes.[35] BTK participated in the regulating of pathways that contribute to rheumatoid arthritis (RA), a multifactorial autoimmune disease.[36] In 2019, Watterson et al reported the discovery of Branebrutinib (60, [Fig. 13]), which is an irreversible BTK inhibitor, and currently in phase II clinical trials for the treatment of RA.[37] It is derived from the earlier described reversible BTK inhibitor BMS-986142 (57, [Fig. 13]) whose quinazolinedione ring system provides two bridging hydrogen bonds with Cys481 and Gln412 through two ordered water molecules.[37] To get a covalent, irreversible BTK inhibitor, they first replaced the quinazolinedione ring with a simple acrylamide acceptor and simplified the hexahydro carbazole core to the 2,3-dimethylindole ring system to achieve compound 58 (BTK IC50 = 0.17 nmol/L, Romas cell IC50 = 16 nmol/L; [Fig. 13]). However, 58 had a poor plasma PK in a mouse model, and this may be possibly due to the high acrylamide reactivity of the compound with thiols such as glutathione. Thus, alternation of accepters makes less contribution to resolving PK issue while sustaining the desirable potency. Therefore, the N-phenylacrylamide linker should be changed. Considering the balance between potency, electrophile reactivity, and PK profile, an S-3-piperidine appended with a but-2-ynamide accepter was introduced into the compound and gave 59 ([Fig. 13]), a covalent BTK inhibitor with acceptable plasma concentrations in vivo but decreased potency (BTK IC50 = 0.52 nmol/L, Romas cell IC50 = 40 nmol/L). At last, a fluoro was introduced at the C5-position of the dimethylindole core and gave branebrutinib (60), which demonstrated improved potency (BTK IC50 = 0.1 nmol/L, Ramos cell IC50 = 7.2 nmol/L) and BTK inactivation rate (3.5 × 10−4 nmol/L−1·min−1). The cocrystal structure of 60 binding to BTK showed that its chiral piperidine linker fit into the cavum of BTK well and the but-2-ynamide accepter formed a covalent bond with Cys481 ([Fig. 14]). Further evaluation showed that the fluorine-substituted analogue 60 had a desirable safety and tolerability profile ([Table 12]), and finally, 60 was advanced for clinical evaluation.
Abbreviations: AUC, area under the concentration–time curve extrapolated to infinity; CL, total clearance; C max, maximum plasma concentration; iv, intravenous injection; MRT, mean residence time; po, oral; T 1/2, elimination half-life; T max, time to reach maximum concentration; V ss, volume at steady state.

Checkpoint kinase 1 (CHK1) is a kind of Ser/Thr protein kinases that mediate the cellular response to DNA damage.[38] CHK1 inhibitors have been identified as potential cancer therapeutics, and had gained substantial interest from both academia and industry. In 2017, Yang et al reported the discovery of a series of CHK1 inhibitors.[38] In their previous work, the compound (61) with a 7-carboxamide thienopyridine core and a piperidine side chain was explored, which had a good potency but suffered from short half-life. Further SAR experiments showed that introducing a methyl at 2-position of the piperidine ring can effectively prolong its half-life. This led to the discovery of 62, a potent CHK1 inhibitor with good potency and PK properties both in vitro and in vivo ([Table 13]).

In the process of identifying a new series of platelet-derived growth factor receptor (PDGFR) inhibitors, Hicken et al reported the discovery of 64, a piperidine-containing PDGFR inhibitor with excellent potency and PK properties.[39] Structure-based design led to the discovery of 63, which had satisfied potency and selectivity. However, 63 suffers from poor oral exposure and bioavailability. Further SAR study of 63 revealed that introducing a fluorine substituent at 3-position of the piperidine ring can effectively improve the PK properties of this series of compounds, which led to the discovery of 64. Compared with 63 which has a symmetrical piperidine ring, 64 has an asymmetric piperidine ring with a fluorine substituent at the 3-position which enables its good oral exposure and bioavailability ([Table 14]).


Abbreviations: AUC, area under the concentration–time curve extrapolated to infinity; CL, total clearance; C max, maximum plasma concentration.
Note: Values were measured in male rat.
Reducing hERG Toxicity
hERG, the abbreviation of “human-ether-a-go-go-related gene,” is a gene encoding a cardiac potassium channel whose protein product is the inner pore-forming part of a key membrane-bound potassium channel in myocardial tissue.[40] When the compound binds to the hERG potassium channel, the outflow of potassium ions is blocked and the repolarization time of cardiomyocytes is prolonged, which may induce a fatal risk of torsades de pointes.[41] [42]
With a wider and deeper knowledge of interaction between hERG inhibitors and their action site, possible strategies for decreasing drugs' hERG affinity have been focused, which include adjusting drug flexibility,[43] changing the charge state,[44] decreasing the pK a,[45] and decreasing the lipophilicity.[45] Originally, the piperidine ring is a kind of structure with high lipophilicity, and usually has higher hERG inhibition than other groups.[46] Introducing a chiral center into the piperidine ring, either changing the position of a substituent or introducing another substituent, would help in decreasing hERG affinity, because by which, the spatial configuration of the compound may be changed, in parallel with a decreased pK a, increased hydrophilicity, as well as modified polarity.
Uprosertib (65, also known as GSK-795) is a kind of protein kinase B (Akt) inhibitor identified by GlaxoSmithKline, and showed activities toward Akt1/2/3.[47] In 2019, Dong et al reported a new series of Akt1 inhibitors, which were based on the structure of uprosertib, and additionally introduced a piperidine moiety.[48] The connection between the primary amine moiety and the α-position of phenyl led to 66, a potent Akt1 inhibitor with an IC50 value of 3 nmol/L but low hERG selectivity. Further optimization was focused on the modification of its piperidine moiety, which led to 67, an Akt1 inhibitor with even better potency and a higher hERG selectivity ([Fig. 15]).

In 2012, Reck et al reported a series of aminopiperidine-containing compounds targeting bacterial type II topoisomerases (DNA gyrase and topo-isomerase IV).[49] In their work, the quinolinone carbonitrile derivative 68 was found to have a potent antibacterial activity, but suffered from hERG inhibition (hERG IC50 = 44 μmol/L) and QT prolongation in vivo. Introducing electron-withdrawing substituents such as hydroxy, methoxy, and fluoro groups at 3-position of the piperidine moiety could reduce the pK a value of this series of compounds, and led to the subsequent hERG inhibition through reduction of binding affinity. Compound 69, a cis-fluoro-substituted analogue, exhibited an improved selectivity over hERG (IC50 = 233 μmol/L) and a maintained potency toward gram-positive organisms ([Fig. 16]). With good inhibition to bacterial growth and high hERG selectivity, 69 was selected as the candidate compound for a deeper preclinical study.

Renin, an aspartic protease, cleaves angiotensinogen to release inactive peptide angiotensin I (Ang-I) and controls the first and rate-limiting step of the renin–angiotensin–aldosterone system.[50] Blockade of renin has been considered as a useful method to treat hypertension and to protect from end-organ damage.[51] In 2014, Ehara et al identified a new class of renin inhibitors.[52] Compound 70, a racemic cis-configured 3,5-disubstituted piperidine with weak renin inhibition, was discovered from a piperidine-based combinatorial library through high-throughput screens. Guided by structure-based design, researchers discovered 71, whose activity was improved a lot but the hERG selectivity was low. Further modification was focused on improving hERG selectivity of this series of compounds. By introducing an (R)-configured hydroxyl at 4-position of the piperidine ring, 72 showed a higher renin inhibition and, most importantly, a good hERG selectivity with a hERG IC50 value of above 30 μmol/L ([Fig. 17]).

Conclusion
Chiral piperidine scaffolds can be used to alter structure patterns with normal orientation, and have fascinated chemists for exploring desired molecules in medicinal chemistry. Due to distinctive three dimensionality that chiral centers impart, chiral piperidine scaffolds usually exhibit good adaptation to the binding site of the protein, resulting in the enhancement in activity and selectivity as well as fewer off-target effects.[53] Besides, π–π stacking interaction of a molecule may be reduced by the introduction of a chiral piperidine ring, thus, drug solubility and PK properties will be improved.[54] Furthermore, chiral piperidine rings were also associated with good hERG selectivity, and this may be due to the modification of polarity and lipophilicity of a molecule.[55] Taken together, chiral piperidine rings are a powerful tool for medicinal chemists to expand novel structures in the current drug discovery.
Conflict of Interest
The authors declare no conflict of interest.
-
Reference
- 1 Laplante SRD, D Fader L, Fandrick KR. et al. Assessing atropisomer axial chirality in drug discovery and development. J Med Chem 2011; 54 (20) 7005-7022
- 2 Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem 2014; 57 (24) 10257-10274
- 3 Silvestri IP, Colbon PJJ. The growing importance of chirality in 3D chemical space exploration and modern drug discovery approaches for Hit-ID: topical innovations. ACS Med Chem Lett 2021; 12 (08) 1220-1229
- 4 Bhutani P, Joshi G, Raja N. et al. U.S. FDA approved drugs from 2015-June 2020: a perspective. J Med Chem 2021; 64 (05) 2339-2381
- 5 Kumorkiewicz-Jamro A, Świergosz T, Sutor K, Spórna-Kucab A, Wybraniec S. Multi-colored shades of betalains: recent advances in betacyanin chemistry. Nat Prod Rep 2021; 38 (12) 2315-2346
- 6 Shan C, Xu J, Cao L. et al. Rapid synthesis of α-chiral piperidines via a highly diastereoselective continuous flow protocol. Org Lett 2022; 24 (17) 3205-3210
- 7 Akamatsu M. Importance of physicochemical properties for the design of new pesticides. J Agric Food Chem 2011; 59 (07) 2909-2917
- 8 Ndungu JM, Krumm SA, Yan D. et al. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase: synthesis, structure-activity relationships, and pharmacokinetics. J Med Chem 2012; 55 (09) 4220-4230
- 9 de Castro Fonseca M, Aguiar CJ, da Rocha Franco JA, Gingold RN, Leite MF. GPR91: expanding the frontiers of Krebs cycle intermediates. Cell Commun Signal 2016; 14: 3
- 10 Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C. Intermediates of metabolism: from bystanders to signalling molecules. Trends Biochem Sci 2016; 41 (05) 460-471
- 11 Tannahill GM, Curtis AM, Adamik J. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013; 496 (7444): 238-242
- 12 Velcicky J, Wilcken R, Cotesta S. et al. Discovery and optimization of novel SUCNR1 inhibitors: design of zwitterionic derivatives with a salt bridge for the improvement of oral exposure. J Med Chem 2020; 63 (17) 9856-9875
- 13 Udvarhelyi A, Rodde S, Wilcken R. ReSCoSS: a flexible quantum chemistry workflow identifying relevant solution conformers of drug-like molecules. J Comput Aided Mol Des 2021; 35 (04) 399-415
- 14 Nadimetla DN, Al Kobaisi M, Bugde ST, Bhosale SV. Tuning achiral to chiral supramolecular helical superstructures. Chem Rec 2020; 20 (08) 793-819
- 15 Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem 2009; 52 (21) 6752-6756
- 16 Wu S, Cai W, Shi Z. et al. Knockdown of MTHFD2 inhibits proliferation and migration of nasopharyngeal carcinoma cells through the ERK signaling pathway. Biochem Biophys Res Commun 2022; 614: 47-55
- 17 Xing M, Yang Y, Huang J. et al. TFPI inhibits breast cancer progression by suppressing ERK/p38 MAPK signaling pathway. Genes Genomics 2022; 44 (07) 801-812
- 18 Poddutoori R, Aardalen K, Aithal K. et al. Discovery of MAP855, an efficacious and selective MEK1/2 inhibitor with an ATP-competitive mode of action. J Med Chem 2022; 65 (05) 4350-4366
- 19 Mainolfi N, Ehara T, Karki RG. et al. Discovery of 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a factor B inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases. J Med Chem 2020; 63 (11) 5697-5722
- 20 Calderon-González KG, Medina-Medina I, Haronikova L. et al. Cryptic in vitro ubiquitin ligase activity of HDMX towards p53 is probably regulated by an induced fit mechanism. Biosci Rep 2022; 42 (07) BSR20220186
- 21 Ma Y, Lahue BR, Gibeau CR. et al. Pivotal role of an aliphatic side chain in the development of an HDM2 inhibitor. ACS Med Chem Lett 2014; 5 (05) 572-575
- 22 Vassilev LT, Vu BT, Graves B. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303 (5659): 844-848
- 23 Rutaganira FU, Barks J, Dhason MS. et al. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and reoccurrence of central nervous system disease by Toxoplasma gondii. J Med Chem 2017; 60 (24) 9976-9989
- 24 Graaf Cd, Donnelly D, Wootten D. et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol Rev 2016; 68 (04) 954-1013
- 25 Decara JM, Vázquez-Villa H, Brea J. et al. Discovery of V-0219: a small-molecule positive allosteric modulator of the glucagon-like peptide-1 receptor toward oral treatment for “diabesity”. J Med Chem 2022; 65 (07) 5449-5461
- 26 Humphreys PG, Bamborough P, Chung CW. et al. Discovery of a potent, cell penetrant, and selective p300/CBP-associated factor (PCAF)/general control nonderepressible 5 (GCN5) bromodomain chemical probe. J Med Chem 2017; 60 (02) 695-709
- 27 Huang L, Li H, Li L. et al. Discovery of pyrrolo[3,2- d]pyrimidin-4-one derivatives as a new class of potent and cell-active inhibitors of P300/CBP-associated factor bromodomain. J Med Chem 2019; 62 (09) 4526-4542
- 28 Lee S, Lee JS. Cellular senescence: a promising strategy for cancer therapy. BMB Rep 2019; 52 (01) 35-41
- 29 Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol 2019; 21 (01) 94-101
- 30 Oh S, Kwon DY, Choi I. et al. Identification of piperidine-3-carboxamide derivatives inducing senescence-like phenotype with antimelanoma activities. ACS Med Chem Lett 2021; 12 (04) 563-571
- 31 Basarab GS, Hill PJ, Garner CE. et al. Optimization of pyrrolamide topoisomerase II inhibitors toward identification of an antibacterial clinical candidate (AZD5099). J Med Chem 2014; 57 (14) 6060-6082
- 32 Karlsson S, Pettersen D, Sörensen H. AZD6564, discovery of a potent 5-substituted isoxazol-3-ol fibrinolysis inhibitor and development of an enantioselective large-scale route for its preparation. ACS Symposium Series 2018; 1307: 151-184
- 33 Shen J, Zhang T, Zhu SJ. et al. Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S). J Med Chem 2019; 62 (15) 7302-7308
- 34 Zhang X, Sheng X, Shen J. et al. Discovery and evaluation of pyrazolo[3,4-d]pyridazinone as a potent and orally active irreversible BTK inhibitor. ACS Med Chem Lett 2019; 11 (10) 1863-1868
- 35 Tichenor MS, Wiener JJM, Rao NL. et al. Discovery of JNJ-64264681: a potent and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem 2022; 65 (21) 14326-14336
- 36 Liu J, Guiadeen D, Krikorian A. et al. Discovery of 8-amino-imidazo[1,5-a]pyrazines as reversible BTK inhibitors for the treatment of rheumatoid arthritis. ACS Med Chem Lett 2015; 7 (02) 198-203
- 37 Watterson SH, Liu Q, Beaudoin Bertrand M. et al. Discovery of Branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton's tyrosine kinase (BTK). J Med Chem 2019; 62 (07) 3228-3250
- 38 Yang B, Vasbinder MM, Hird AW. et al. Adventures in scaffold morphing: discovery of fused ring heterocyclic checkpoint kinase 1 (CHK1) inhibitors. J Med Chem 2018; 61 (03) 1061-1073
- 39 Hicken EJ, Marmsater FP, Munson MC. et al. Discovery of a novel class of imidazo[1,2-a]pyridines with potent PDGFR activity and oral bioavailability. ACS Med Chem Lett 2013; 5 (01) 78-83
- 40 Zhang X, Mao J, Wei M, Qi Y, Zhang JZH. HergSPred: accurate classification of hERG blockers/nonblockers with machine-learning models. J Chem Inf Model 2022; 62 (08) 1830-1839
- 41 Asahi Y, Nomura F, Abe Y. et al. Electrophysiological evaluation of pentamidine and 17-AAG in human stem cell-derived cardiomyocytes for safety assessment. Eur J Pharmacol 2019; 842: 221-230
- 42 Sharifi M. Computational approaches to understand the adverse drug effect on potassium, sodium and calcium channels for predicting TdP cardiac arrhythmias. J Mol Graph Model 2017; 76: 152-160
- 43 Blum CA, Zheng X, De Lombaert S. Design, synthesis, and biological evaluation of substituted 2-cyclohexyl-4-phenyl-1H-imidazoles: potent and selective neuropeptide Y Y5-receptor antagonists. J Med Chem 2004; 47 (09) 2318-2325
- 44 Rampe D, Wible B, Brown AM, Dage RC. Effects of terfenadine and its metabolites on a delayed rectifier K+ channel cloned from human heart. Mol Pharmacol 1993; 44 (06) 1240-1245
- 45 Ganellin R, Roberts S, Jefferies R. Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies. Amsterdam: Academic Press; 2013
- 46 Tschirhart JN, Zhang S. Fentanyl-induced block of hERG channels is exacerbated by hypoxia, hypokalemia, alkalosis, and the presence of hERG1b. Mol Pharmacol 2020; 98 (04) 508-517
- 47 Uko NE, Güner OF, Matesic DF, Bowen JP. Akt pathway inhibitors. Curr Top Med Chem 2020; 20 (10) 883-900
- 48 Dong X, Zhan W, Zhao M. et al. Discovery of 3,4,6-trisubstituted piperidine derivatives as orally active, low hERG blocking Akt inhibitors via conformational restriction and structure-based design. J Med Chem 2019; 62 (15) 7264-7288
- 49 Reck F, Alm RA, Brassil P. et al. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pK(a): antibacterial agents with an improved safety profile. J Med Chem 2012; 55 (15) 6916-6933
- 50 Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov 2002; 1 (08) 621-636
- 51 Skeggs Jr LT, Kahn JR, Lentz K, Shumway NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med 1957; 106 (03) 439-453
- 52 Ehara T, Irie O, Kosaka T. et al. Structure-based design of substituted piperidines as a new class of highly efficacious oral direct Renin inhibitors. ACS Med Chem Lett 2014; 5 (07) 787-792
- 53 Gao Y, Zhao X, Sun X. et al. Enantioselective detection, bioactivity, and degradation of the novel chiral fungicide oxathiapiprolin. J Agric Food Chem 2021; 69 (11) 3289-3297
- 54 Saha D, Kharbanda A, Yan W, Lakkaniga NR, Frett B, Li HY. The exploration of chirality for improved druggability within the human kinome. J Med Chem 2020; 63 (02) 441-469
- 55 Feng PF, Zhang B, Zhao L. et al. Intracellular mechanism of rosuvastatin-induced decrease in mature hERG protein expression on membrane. Mol Pharm 2019; 16 (04) 1477-1488
Address for correspondence
Publication History
Received: 28 July 2022
Accepted: 26 January 2023
Article published online:
15 March 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
Reference
- 1 Laplante SRD, D Fader L, Fandrick KR. et al. Assessing atropisomer axial chirality in drug discovery and development. J Med Chem 2011; 54 (20) 7005-7022
- 2 Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem 2014; 57 (24) 10257-10274
- 3 Silvestri IP, Colbon PJJ. The growing importance of chirality in 3D chemical space exploration and modern drug discovery approaches for Hit-ID: topical innovations. ACS Med Chem Lett 2021; 12 (08) 1220-1229
- 4 Bhutani P, Joshi G, Raja N. et al. U.S. FDA approved drugs from 2015-June 2020: a perspective. J Med Chem 2021; 64 (05) 2339-2381
- 5 Kumorkiewicz-Jamro A, Świergosz T, Sutor K, Spórna-Kucab A, Wybraniec S. Multi-colored shades of betalains: recent advances in betacyanin chemistry. Nat Prod Rep 2021; 38 (12) 2315-2346
- 6 Shan C, Xu J, Cao L. et al. Rapid synthesis of α-chiral piperidines via a highly diastereoselective continuous flow protocol. Org Lett 2022; 24 (17) 3205-3210
- 7 Akamatsu M. Importance of physicochemical properties for the design of new pesticides. J Agric Food Chem 2011; 59 (07) 2909-2917
- 8 Ndungu JM, Krumm SA, Yan D. et al. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase: synthesis, structure-activity relationships, and pharmacokinetics. J Med Chem 2012; 55 (09) 4220-4230
- 9 de Castro Fonseca M, Aguiar CJ, da Rocha Franco JA, Gingold RN, Leite MF. GPR91: expanding the frontiers of Krebs cycle intermediates. Cell Commun Signal 2016; 14: 3
- 10 Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C. Intermediates of metabolism: from bystanders to signalling molecules. Trends Biochem Sci 2016; 41 (05) 460-471
- 11 Tannahill GM, Curtis AM, Adamik J. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013; 496 (7444): 238-242
- 12 Velcicky J, Wilcken R, Cotesta S. et al. Discovery and optimization of novel SUCNR1 inhibitors: design of zwitterionic derivatives with a salt bridge for the improvement of oral exposure. J Med Chem 2020; 63 (17) 9856-9875
- 13 Udvarhelyi A, Rodde S, Wilcken R. ReSCoSS: a flexible quantum chemistry workflow identifying relevant solution conformers of drug-like molecules. J Comput Aided Mol Des 2021; 35 (04) 399-415
- 14 Nadimetla DN, Al Kobaisi M, Bugde ST, Bhosale SV. Tuning achiral to chiral supramolecular helical superstructures. Chem Rec 2020; 20 (08) 793-819
- 15 Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem 2009; 52 (21) 6752-6756
- 16 Wu S, Cai W, Shi Z. et al. Knockdown of MTHFD2 inhibits proliferation and migration of nasopharyngeal carcinoma cells through the ERK signaling pathway. Biochem Biophys Res Commun 2022; 614: 47-55
- 17 Xing M, Yang Y, Huang J. et al. TFPI inhibits breast cancer progression by suppressing ERK/p38 MAPK signaling pathway. Genes Genomics 2022; 44 (07) 801-812
- 18 Poddutoori R, Aardalen K, Aithal K. et al. Discovery of MAP855, an efficacious and selective MEK1/2 inhibitor with an ATP-competitive mode of action. J Med Chem 2022; 65 (05) 4350-4366
- 19 Mainolfi N, Ehara T, Karki RG. et al. Discovery of 4-((2S,4S)-4-ethoxy-1-((5-methoxy-7-methyl-1H-indol-4-yl)methyl)piperidin-2-yl)benzoic acid (LNP023), a factor B inhibitor specifically designed to be applicable to treating a diverse array of complement mediated diseases. J Med Chem 2020; 63 (11) 5697-5722
- 20 Calderon-González KG, Medina-Medina I, Haronikova L. et al. Cryptic in vitro ubiquitin ligase activity of HDMX towards p53 is probably regulated by an induced fit mechanism. Biosci Rep 2022; 42 (07) BSR20220186
- 21 Ma Y, Lahue BR, Gibeau CR. et al. Pivotal role of an aliphatic side chain in the development of an HDM2 inhibitor. ACS Med Chem Lett 2014; 5 (05) 572-575
- 22 Vassilev LT, Vu BT, Graves B. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303 (5659): 844-848
- 23 Rutaganira FU, Barks J, Dhason MS. et al. Inhibition of calcium dependent protein kinase 1 (CDPK1) by pyrazolopyrimidine analogs decreases establishment and reoccurrence of central nervous system disease by Toxoplasma gondii. J Med Chem 2017; 60 (24) 9976-9989
- 24 Graaf Cd, Donnelly D, Wootten D. et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol Rev 2016; 68 (04) 954-1013
- 25 Decara JM, Vázquez-Villa H, Brea J. et al. Discovery of V-0219: a small-molecule positive allosteric modulator of the glucagon-like peptide-1 receptor toward oral treatment for “diabesity”. J Med Chem 2022; 65 (07) 5449-5461
- 26 Humphreys PG, Bamborough P, Chung CW. et al. Discovery of a potent, cell penetrant, and selective p300/CBP-associated factor (PCAF)/general control nonderepressible 5 (GCN5) bromodomain chemical probe. J Med Chem 2017; 60 (02) 695-709
- 27 Huang L, Li H, Li L. et al. Discovery of pyrrolo[3,2- d]pyrimidin-4-one derivatives as a new class of potent and cell-active inhibitors of P300/CBP-associated factor bromodomain. J Med Chem 2019; 62 (09) 4526-4542
- 28 Lee S, Lee JS. Cellular senescence: a promising strategy for cancer therapy. BMB Rep 2019; 52 (01) 35-41
- 29 Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol 2019; 21 (01) 94-101
- 30 Oh S, Kwon DY, Choi I. et al. Identification of piperidine-3-carboxamide derivatives inducing senescence-like phenotype with antimelanoma activities. ACS Med Chem Lett 2021; 12 (04) 563-571
- 31 Basarab GS, Hill PJ, Garner CE. et al. Optimization of pyrrolamide topoisomerase II inhibitors toward identification of an antibacterial clinical candidate (AZD5099). J Med Chem 2014; 57 (14) 6060-6082
- 32 Karlsson S, Pettersen D, Sörensen H. AZD6564, discovery of a potent 5-substituted isoxazol-3-ol fibrinolysis inhibitor and development of an enantioselective large-scale route for its preparation. ACS Symposium Series 2018; 1307: 151-184
- 33 Shen J, Zhang T, Zhu SJ. et al. Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S). J Med Chem 2019; 62 (15) 7302-7308
- 34 Zhang X, Sheng X, Shen J. et al. Discovery and evaluation of pyrazolo[3,4-d]pyridazinone as a potent and orally active irreversible BTK inhibitor. ACS Med Chem Lett 2019; 11 (10) 1863-1868
- 35 Tichenor MS, Wiener JJM, Rao NL. et al. Discovery of JNJ-64264681: a potent and selective covalent inhibitor of Bruton's tyrosine kinase. J Med Chem 2022; 65 (21) 14326-14336
- 36 Liu J, Guiadeen D, Krikorian A. et al. Discovery of 8-amino-imidazo[1,5-a]pyrazines as reversible BTK inhibitors for the treatment of rheumatoid arthritis. ACS Med Chem Lett 2015; 7 (02) 198-203
- 37 Watterson SH, Liu Q, Beaudoin Bertrand M. et al. Discovery of Branebrutinib (BMS-986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton's tyrosine kinase (BTK). J Med Chem 2019; 62 (07) 3228-3250
- 38 Yang B, Vasbinder MM, Hird AW. et al. Adventures in scaffold morphing: discovery of fused ring heterocyclic checkpoint kinase 1 (CHK1) inhibitors. J Med Chem 2018; 61 (03) 1061-1073
- 39 Hicken EJ, Marmsater FP, Munson MC. et al. Discovery of a novel class of imidazo[1,2-a]pyridines with potent PDGFR activity and oral bioavailability. ACS Med Chem Lett 2013; 5 (01) 78-83
- 40 Zhang X, Mao J, Wei M, Qi Y, Zhang JZH. HergSPred: accurate classification of hERG blockers/nonblockers with machine-learning models. J Chem Inf Model 2022; 62 (08) 1830-1839
- 41 Asahi Y, Nomura F, Abe Y. et al. Electrophysiological evaluation of pentamidine and 17-AAG in human stem cell-derived cardiomyocytes for safety assessment. Eur J Pharmacol 2019; 842: 221-230
- 42 Sharifi M. Computational approaches to understand the adverse drug effect on potassium, sodium and calcium channels for predicting TdP cardiac arrhythmias. J Mol Graph Model 2017; 76: 152-160
- 43 Blum CA, Zheng X, De Lombaert S. Design, synthesis, and biological evaluation of substituted 2-cyclohexyl-4-phenyl-1H-imidazoles: potent and selective neuropeptide Y Y5-receptor antagonists. J Med Chem 2004; 47 (09) 2318-2325
- 44 Rampe D, Wible B, Brown AM, Dage RC. Effects of terfenadine and its metabolites on a delayed rectifier K+ channel cloned from human heart. Mol Pharmacol 1993; 44 (06) 1240-1245
- 45 Ganellin R, Roberts S, Jefferies R. Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies. Amsterdam: Academic Press; 2013
- 46 Tschirhart JN, Zhang S. Fentanyl-induced block of hERG channels is exacerbated by hypoxia, hypokalemia, alkalosis, and the presence of hERG1b. Mol Pharmacol 2020; 98 (04) 508-517
- 47 Uko NE, Güner OF, Matesic DF, Bowen JP. Akt pathway inhibitors. Curr Top Med Chem 2020; 20 (10) 883-900
- 48 Dong X, Zhan W, Zhao M. et al. Discovery of 3,4,6-trisubstituted piperidine derivatives as orally active, low hERG blocking Akt inhibitors via conformational restriction and structure-based design. J Med Chem 2019; 62 (15) 7264-7288
- 49 Reck F, Alm RA, Brassil P. et al. Novel N-linked aminopiperidine inhibitors of bacterial topoisomerase type II with reduced pK(a): antibacterial agents with an improved safety profile. J Med Chem 2012; 55 (15) 6916-6933
- 50 Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov 2002; 1 (08) 621-636
- 51 Skeggs Jr LT, Kahn JR, Lentz K, Shumway NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med 1957; 106 (03) 439-453
- 52 Ehara T, Irie O, Kosaka T. et al. Structure-based design of substituted piperidines as a new class of highly efficacious oral direct Renin inhibitors. ACS Med Chem Lett 2014; 5 (07) 787-792
- 53 Gao Y, Zhao X, Sun X. et al. Enantioselective detection, bioactivity, and degradation of the novel chiral fungicide oxathiapiprolin. J Agric Food Chem 2021; 69 (11) 3289-3297
- 54 Saha D, Kharbanda A, Yan W, Lakkaniga NR, Frett B, Li HY. The exploration of chirality for improved druggability within the human kinome. J Med Chem 2020; 63 (02) 441-469
- 55 Feng PF, Zhang B, Zhao L. et al. Intracellular mechanism of rosuvastatin-induced decrease in mature hERG protein expression on membrane. Mol Pharm 2019; 16 (04) 1477-1488