Discussion
Terminology and Classification
Spina bifida: The term spina bifida classically refers to the defective fusion of posterior spinal bony elements. The spectrum of cases in the study included bony defects in the anterior arch as well.
Neural placode: Segment of non-neurulated embryonic neural tissue, which is arrested at the neural plate stage.
Open and closed spinal dysraphism: The neural placode is exposed through a skin defect in the midline in an open spinal dysraphism. There is no exposed neural tissue in the case of a closed spinal dysraphism.
Posterior spinal dysraphism: These are defects of the laminae and the spinous process of the vertebrae and/or the intervening mesenchymal elements.
Anterior spinal dysraphism: These are defects in the vertebral body/pedicles/neural foramina of the vertebrae and/or the intervening mesenchymal elements.
Dysraphism is confined to the spinal canal with or without a dural defect.
Primary defects involving multiple (more than 2) anatomical compartments: (prevertebral mesenchyme, anterior spinal bony elements, spinal canal, posterior spinal bony elements, postvertebral mesenchyme, and other organ systems)
Nondysraphic masses of the spinal region: Tumors with a close association with the notochord and the neural tube with neurulation remaining unaffected.
Cutaneous Stigmata of Spinal Dysraphism: Skin Predicts the Spine
Larger defects are usually obvious, whereas small or occult malformations causing tethering of the cord may be asymptomatic at birth. Also, 69.1% of patients in our study had a cutaneous marker of spinal dysraphism, most commonly in the lumbosacral region. Hence, the first steps in diagnosis are these clinical cues such as the presence or absence of an exposed neural placode, the presence of a subcutaneous mass, or a dermal sinus in case of closed dysraphisms. A combination of cutaneous markers is possible ([Table 3]). A low-lying (<2.5 cm from anal verge) sacral dimple may not always be associated with underlying spinal dysraphism ([Table 4]).[7]
Table 3
Skin Stigmata and commonly associated conditions
|
Cutaneous marker
|
Associated condition
|
|
Hypertrichosis (Faun tail)
|
Split cord malformation
|
|
Sacral dimple
|
Dorsal dermal sinus
|
|
Subcutaneous lipoma
|
Lipomyelomeningocele
|
|
Absent or asymmetric gluteal cleft
|
Sacral agenesis
|
|
Others
|
Non-specific occult dysraphism
|
Table 4
Skin stigmata of occult spinal dysraphism
|
High probability of underlying dysraphism
|
Low probability
|
|
Hypertrichosis (Faun tail nevus)
Dimples (atypical large dimples > 2.5 cm from the anal verge)
Acrochordons/pseudotails/true tails
Subcutaneous lipomas
Hemangioma
Aplasia cutis or scar
Dermoid cyst or sinus
|
Capillary malformation (port-wine stain)
Telangiectasia
Hyperpigmentation
Melanocytic nevi
Low-lying sacral dimple(<2.5 cm from the anal verge)
|
An embryological knowledge of these complex interlinked processes, although necessary, is often confusing while arriving at a diagnosis. Hence, the need for a simpler and more practical anatomical–neuroradiological classification ([Table 5]).
Table 5
Anatomical–clinicoradiological classification of spinal dysraphism (Stanley Hospital classification)
|
1)Posterior spinal dysraphism
Posterior: Open spinal dysraphism:
Myelomeningocele
Myelocele
Hemi myelomeningocele
Hemi myelocele
Posterior: Closed spinal dysraphism
With subcutaneous mass:
Meningocele
Saccular limited dorsal myeloschisis
Myelocystocele
Posterior lipomyelocele (spinal lipoma Types IB, IIB, IIIB,)
Posterior lipomyelomeningocele (spinal lipoma type VB)
Without subcutaneous mass:
With cutaneous stigmata
other than a dermal sinus: Nonsaccular limited dorsal myeloschisis
Post vertebral neurenteric cysts
With dermal sinus: Dorsal dermal sinus
2)Anterior spinal dysraphism:
Anterior myelomeningocele
Anterior meningocele
Anterior myelocele
Anterior myelocystocele
Anterior lipomyelocele (spinal lipoma type IC, IIC, IIIC)
Anterior lipomyelomeningocele (spinal lipoma type VA)
Intrasacral meningocele
Prevertebral neurenteric cysts
3)Dysraphism confined to the spinal canal:
Intramedullary spinal lipoma
Intradural lipomas
Type Ia,
Type IIa, IId
Type IIIa
Type IV–IVA-apical, IVB: mid filar and IV C: filar tip Lipomas
Tight filum terminale (type IVD)
Intrasacral meningocele
Persistent terminal ventricle
Retained medullary cord
Split cord malformations
Types 1 and 2 diastematomyelia
Composite type diastematomyelia
Partial type diastematomyelia
Neuroenteric cysts
(Epi)dermoid/dermoid
Segmental spinal dysgenesis: type 1
4) Dysraphism involving multiple anatomical compartments
Dorsal enteric fistula
Vertebral duplication
Caudal regression syndrome
Segmental spinal dysgenesis: type 2
OEIS, VACTERL, Currarino's triad associations
5) Nondysraphic masses of the spinal region
Sacrococcygeal teratoma
Sacral chordoma
|
A Review of the Anomalies and Associated Findings
Myelomeningocele and Myelocele: These are classified under open dysraphisms. The most common type of open spinal dysraphism is myelomeningocele ([Fig. 9]). The neural placode protrudes above the skin surface in case of a myelomeningocele due to cystic dilatation of subarachnoid space whereas a myelocele does not protrude above the skin surface.
 Fig. 9 Posterior myelomeningocele. (A) Image shows neural tissue and CSF directly exposed to the environment in the lumbar region. (B–D) T2 axial, sagittal images, and illustration show the spinal cord herniating in a CSF-filled sac (arrows) through a defect in the posterior vertebral elements (arrowheads).
Fig. 9 Posterior myelomeningocele. (A) Image shows neural tissue and CSF directly exposed to the environment in the lumbar region. (B–D) T2 axial, sagittal images, and illustration show the spinal cord herniating in a CSF-filled sac (arrows) through a defect in the posterior vertebral elements (arrowheads).
Most are located at the lumbosacral level, and the placode is terminal (below the S2 level). A midline skin defect causes the external surface of the placode to be directly visible on inspection.
The nerve roots originate from this surface and course obliquely through the subarachnoid space to reach their corresponding neural foramina. Because both are exposed to the skin surface, ulceration and risk of infection are more, which increases the mortality rate. Hence, surgery is needed soon after birth or even before birth with exit. Preoperative imaging with MRI helps in better anatomic characterization and assessing the associated abnormalities Chiari malformations, syrinx, hydrocephalus, and split cord malformations. In the proposed classification, meningoceles and myelomeningoceles have been further classified into those extending anteriorly and those extending posteriorly.
Myelomeningocele is almost always associated with Chiari 2 malformation.
McLone and Knepper[13] proposed a theory to explain this consistent association. CSF leaks freely through the spinal defect into the amniotic sac because the neural tube remains non-neurulated. The resulting chronic CSF hypotension causes failure of expansion of the rhombencephalic vesicle, causing a lack of induction of the perineural mesenchyme of the posterior cranial fossa. Both the cerebellum and brain stem eventually are forced to develop within a smaller than normal posterior fossa and consequently herniate through the tentorial groove and the foramen magnum.
Hemimyelocele and Hemimyelomeningocele
These are extremely rare. These occur when a myelomeningocele or myelocele is associated with diastematomyelia and one hemicord fails to neurulate.
Meningocele
Meningoceles are herniation of CSF-filled sacs lined by dura and arachnoid. When it occurs through a posterior bony spina bifida, they are called posterior meningoceles. The most common are lumbar or sacral posterior meningocele ([Fig. 10]); therefore, thoracic and even cervical meningoceles may be found. The spinal cord is not contained within the meningocele although it may be tethered to its neck. Most of these are extradural in location, causing compression of the bony sacral canal. A few reported meningoceles may be a dome stalk variety of saccular limited dorsal myeloschisis.[8]
 Fig. 10 Posterior meningocele. (A) Clinical image showing the skin covered swelling in the lumbar region. (B) Myelogram, (C) T2 sagittal, and (D) T2 axial images, and (E) illustration shows CSF-filled sac herniating through a defect in the posterior vertebral elements. There is no neural tissue within the herniated sac.
Fig. 10 Posterior meningocele. (A) Clinical image showing the skin covered swelling in the lumbar region. (B) Myelogram, (C) T2 sagittal, and (D) T2 axial images, and (E) illustration shows CSF-filled sac herniating through a defect in the posterior vertebral elements. There is no neural tissue within the herniated sac.
Anterior Neural Tube Defects
Myelocele, myelmeningoceles, myelocystocele, meningoceles, and lipomyelocoeles usually occur posteriorly. When these lesions occur anteriorly or laterally with defects in the vertebral body, pedicles or neural foraminae, they are collectively called anterior neural tube defects. Golden and Chernoff hypothesized multiple sites of anterior neural tube closure and that anterior neural tube defects occur either due to failure of closure to occur or from the failure of two closures to meet. Another possibility would be due to CSF pulsation resulting in herniation through anteriorly located cleft in the vertebra.
Anterior Myelocystocele
Herniation of dilated cord with syrinx (hydro melic cord) through anterior vertebral defect is called anterior myelocystocele ([Fig. 11]). Meningocele requires a simple surgical repair, whereas myelocystocele requires untethering of cord, decompression of syrinx along with excision, and repair of myelocystocele.[25]
 Fig. 11 Anterior thoracic myelocystocele. (A) T2 axial and (D) T2 coronal images with (C) myelogram and (B) axial CT section showing an anterior vertebral defect with the herniation of a hydromelic dilated cord into a CSF-filled sac anterolaterally.
Fig. 11 Anterior thoracic myelocystocele. (A) T2 axial and (D) T2 coronal images with (C) myelogram and (B) axial CT section showing an anterior vertebral defect with the herniation of a hydromelic dilated cord into a CSF-filled sac anterolaterally.
Anterior Myelomeningocele
Posterior myelomeningoceles are more common in the lumbosacral region (80%). Anterior myelomeningoceles ([Fig. 12]) are rarely seen in the sacral region and very rarely seen in the thoracic/cervical region. The embryology behind this is an incomplete development of mesoderm around the neural tube, which results in the anterior defect through which there is herniation of meninges and other contents.
 Fig. 12 Anterior herniation of the cord (anterior myelomeningocele). (A) Myelogram, (B) T2 sagittal and (C) T2 coronal images showing an anterior vertebral defect with herniation of the cord and a CSF-filled sac anteriorly into the prevertebral space. There is also a neurenteric cyst noted in the myelogram (A).
Fig. 12 Anterior herniation of the cord (anterior myelomeningocele). (A) Myelogram, (B) T2 sagittal and (C) T2 coronal images showing an anterior vertebral defect with herniation of the cord and a CSF-filled sac anteriorly into the prevertebral space. There is also a neurenteric cyst noted in the myelogram (A).
Multiple lateral thoracic meningoceles are usually associated with neurofibromatosis (85%). Patients may present with sudden breathlessness secondary to rupture of meningocele resulting in hydrothorax.
Myelocystocele
Myelocystoceles occur due to herniation of dilated central canal forming terminal syringohydromyelia (syringocele) through a posterior ([Fig. 13]) or anterior vertebral defect into an expanded CSF-filled dural sheath (meningocele). It usually results from defective secondary neurulation that affects the CSF flow dynamics. The inner terminal syrinx communicates with the central canal of the spinal cord and the outer meningocele is continuous with the spinal subarachnoid space. The syringocele and meningocele usually do not communicate with each other.[14] Although classified as terminal and non-terminal myelocystocles earlier, many of the “non terminal” myelocystocles were found to be the saccular type of limited dorsal myeloschisis. Thus, it is recommended to stick to the term “myelocystocele (anterior/posterior).
 Fig. 13 Posterior myelocystocele. (A) Image shows skin-covered swelling in the lumbar region. (B) Myelogram, (C) T2 axial, (D) and T1 sagittal images and (E) illustration shows the cord with a dilated central canal (syrinx, arrowheads) within the CSF-containing sac (arrows), which has herniated posteriorly through a vertebral defect.
Fig. 13 Posterior myelocystocele. (A) Image shows skin-covered swelling in the lumbar region. (B) Myelogram, (C) T2 axial, (D) and T1 sagittal images and (E) illustration shows the cord with a dilated central canal (syrinx, arrowheads) within the CSF-containing sac (arrows), which has herniated posteriorly through a vertebral defect.
Spinal Lipomas
The embryonic genesis of spinal lipoma cases is marked by a premature disjunction between the neural and cutaneous ectoderm during the process of primary neurulation. Embryologists often consider closed caudal spinal lesions to be the result of failed secondary neurulation. But still, some confusion in terms of the embryogenesis of spinal lipomas remains.
Spinal lipomas: Embryologically, Type I is defined as pure primary neurulation failure; Type II ranges from primary to secondary neurulation failure; Type III consists of secondary neurulation failure (early phase); and Type IV is defined as secondary neurulation failure (late phase)[12]. This classification of spinal lipomas based on the embryonic stage has the potential for clinical use and agrees well with both clinical and surgical findings.[12] However, we have now added subtypes to include the extension of the lipoma posteriorly (IB/IIB/IIB) and anteriorly (IC/IIC/IIIC) respectively as lipomyeloceles to add on to the previous classification. The rare intramedullary lipomas could be classified as type VI Spinal lipomas ([Fig. 17]) ([Table 6]).
Table 6
Spinal lipomas ([Fig. 20])
|
Type I
|
Intradural lipomas above the level of Conus (Lipoma: placode interface within the dura)
|
|
IA: Those lipomas that are dorsal to the cord or extend cranially/caudally/around the spinal cord; entirely confined to the intradural space
|
|
IB: Dorsal extension as a lipomyelocele
|
|
IC: Ventral extension as a lipomyelocele
|
|
Type II
|
Transitional and chaotic lipomas
|
|
IIA: Transitional lipoma above the last dorsal root entry zone of the conus
|
|
IIB: Dorsal extension as a lipomyelocele
|
|
IIC: Ventral extension as a lipomyelocele
|
|
II D: Involving ventral fat and entangling the nerve roots: chaotic lipoma
|
|
Type III
|
Conal lipomas arising from the level of conus medullaris below the level of last dorsal root entry zone
|
|
III A: Confined to intradural space
|
|
IIIB: Dorsal extension as a lipomyelocele
|
|
IIIC- Ventral extension as a lipomyelocele
|
|
Type IV
|
Filar lipomas: Lipomas of the filum terminale
|
|
IV A: Apical lipoma
|
|
IV B: Mid filar lipoma
|
|
IV C: Filar tip lipoma
|
|
IV D: Tight filum terminale
|
|
Type V
|
Lipomyelomeningocele: Lipoma- placode interface outside the spinal canal
|
|
V A- Anterior lipomyelomeningocele
|
|
V B- Posterior lipomyelomeningocele
|
|
Type VI
|
Intramedullary lipoma
|
Type I: Primary Neurulation Failure
Type I spinal lipoma is one in which the lipoma–cord interface occurs on the dorsal surface of the spinal cord (Type IA), while the conus medullaris is located distally or ventrally with respect to the caudal end of the lipoma. It may be found extending dorsally (Type IB) or ventrally (Type IC) through a spinal defect as lipomyeloceles and be associated with a tethered cord. They are usually seen in the lumbosacral region. Large lipomas may cause cord displacement. On MRI, lipomas follow the signal intensity of subcutaneous fat on all sequences ([Fig. 14]).
 Fig. 14 Lipoma, Types I and II. (A) T2 sagittal image showing an intraspinal lipoma attached to the dorsal cord above the conus medullaris: this is type I. Illustrations show type (B) IA, (C) IB, and (C) IC variants. (E) T1 sagittal image shows a lipoma attached to the cauda equina: Type II. Illustrations show type (F) IIA, (G) IIB, and (H) IIC.
Fig. 14 Lipoma, Types I and II. (A) T2 sagittal image showing an intraspinal lipoma attached to the dorsal cord above the conus medullaris: this is type I. Illustrations show type (B) IA, (C) IB, and (C) IC variants. (E) T1 sagittal image shows a lipoma attached to the cauda equina: Type II. Illustrations show type (F) IIA, (G) IIB, and (H) IIC.
Type II: Failed Neurulation Between the Primary and Secondary Stages
Type II spinal lipomas include the transitional and chaotic lipomas that have a clearly discernible demarcation with the cord surface cranially but caudally either involve the conus medullaris, entangle the nerve roots of the cauda equina, or along with unpredictable involvement of the ventral fat as in the case of chaotic lipomas (Type IID). They may extend dorsally (Type IIB) or ventrally as lipomyeloceles too (Type IIC) ([Fig. 14]).
The Type I spinal lipomas are formed purely by failed primary neurulation; the conus medullaris is left unaffected. The tapered shape of the end of the spinal cord suggests the presence of the conus medullaris. In contrast, a sharp tapered end to the spinal cord is not clearly discernible in Type II spinal lipomas. Dural lipomatosis occurring due to factors such as steroid abuse is a differential to be considered in the case of diffuse fatty proliferation.
Type III: Early Phase Secondary Neurulation Failure
This type of spinal lipoma involves the conus medullaris and is always distal to the origin of the last branch of the spinal nerves on the cord. The caudal end of the conus is unformed and directly connects to the spinal lipoma. The lipoma extends in the caudal direction and usually remains intradural (Type IIIa). Earlier classifications propounded that in contrast to Type II, in Type III spinal lipomas, there is never a pathological spina bifida or dural defect due to the lipoma. A few of our cases showed extension of these caudal lipomas posteriorly and anteriorly as lipomyeloceles and hence, we have added types IIIb and IIIc, respectively ([Fig. 15]), to indicate this extension.
 Fig. 15 Conal (Type III) and filar (Type IV) lipomas. (A) T1 sagittal image showing a tethered cord with a lipoma attached to the conus of the cord: conal lipoma. Illustrations show type (B) IIIA, (C) IIIB, and (D) IIIC variants. (E) The location of dorsal (arrow), transitional (star, dotted lines indicate chaotic variant), and conal (arrowhead) lipomas. (F) T1 sagittal image shows a lipoma attached to the apex of the cord and filum terminale: apical filar lipoma. (G) T2 sagittal image shows a hydromelic low-lying cord with a lipoma attached to the tip of the filum terminale: terminal lipoma. (H) Type IVA: apical filar lipoma, (I) Type IVB: mid filar lipoma, (J) Type IVC: filar tip lipoma.
Fig. 15 Conal (Type III) and filar (Type IV) lipomas. (A) T1 sagittal image showing a tethered cord with a lipoma attached to the conus of the cord: conal lipoma. Illustrations show type (B) IIIA, (C) IIIB, and (D) IIIC variants. (E) The location of dorsal (arrow), transitional (star, dotted lines indicate chaotic variant), and conal (arrowhead) lipomas. (F) T1 sagittal image shows a lipoma attached to the apex of the cord and filum terminale: apical filar lipoma. (G) T2 sagittal image shows a hydromelic low-lying cord with a lipoma attached to the tip of the filum terminale: terminal lipoma. (H) Type IVA: apical filar lipoma, (I) Type IVB: mid filar lipoma, (J) Type IVC: filar tip lipoma.
Type IV: Late Phase Secondary Neurulation Failure
In this type, the spinal lipoma is located in the filum terminale. The caudal end of the conus medullaris is clearly discernible ([Fig. 15]). On imaging filar lipoma appears hyperintense on T1- and T2-weighted images.
Lipomas of the filum terminale could be further classified ([Fig. 15]) as Type IVA: abutting the apex of the conus medullaris; apical lipoma that requires meticulous dissection during surgery; Type IVB: mid filar lipomas with normal-appearing/thickened filum terminale cranially and caudally; and Type IVC: filar tip lipomas that were always associated with a low-lying cord and tethered cord syndromes among the cases analyzed.
Tight Filum Terminale (Type IVD Lipoma)
It is characterized by shortening and hypertrophy of filum terminale that cause tethering of cord and impairs the ascent of conus medullaris. Embryologically, the defect lies in retrogressive differentiation during secondary neurulation.
On imaging, it is characterized by a thick filum terminale ([Fig. 16]) due to fibrous or fatty infiltration (thickness measuring > 2 mm at L5–S1) and a low-lying conus medullaris.
 Fig. 16 Variability in the ending of the cord. (A) Normal termination of the cord, with a thin filum. (B) Thick filum terminale. (C) Cyst of the filum terminale. (D) Lipoma of the filum terminale.
Fig. 16 Variability in the ending of the cord. (A) Normal termination of the cord, with a thin filum. (B) Thick filum terminale. (C) Cyst of the filum terminale. (D) Lipoma of the filum terminale.
Also, 1.5% to 5% of the normal adult population may show fat within filum terminale on MRI and hence the finding is considered a normal variant unless it is associated with tethered cord syndrome.[15]
[16]
Lipomyelocele and Lipomyelomeningocele
Lipomyeloceles and lipomyelomeningoceles are characterized by a subcutaneous fatty mass located above the intergluteal crease and usually extending asymmetrically into one buttock.
In a lipomyelocele (lipomyeloschisis), the placode–lipoma interface lies within the spinal canal ([Fig. 17]) and may extend over several vertebral levels. There is continuity of the intraspinal lipoma with the subcutaneous fat through a posterior bony spina bifida.[17] Any of the types I, II, and III spinal lipomas may present as a lipomyelocele.
 Fig. 17 Posterior lipomyelocele. (A) Image showing a skin-covered swelling in the lumbar region. (B) T2 axial and (C) T1 sagittal images and illustration (D) showing an intraspinal lipoma herniating into the subcutaneous fat through a posterior spina bifida.
Fig. 17 Posterior lipomyelocele. (A) Image showing a skin-covered swelling in the lumbar region. (B) T2 axial and (C) T1 sagittal images and illustration (D) showing an intraspinal lipoma herniating into the subcutaneous fat through a posterior spina bifida.
In lipomyelomeningoceles, the placode–lipoma interface lies outside the spinal canal because of expansion of the underlying subarachnoid spaces, resulting in a posterior meningocele. Anterior and posterior lipomyelomeningocoeles can be classified as Va and Vb, respectively, ([Fig. 19]).
 Fig. 18 Intramedullary lipoma and dural lipomatosis (A) T2 sagittal image showing an intramedullary lipoma expanding the cord: Type VI. (B) T1 sagittal image showing an intramedullary lipoma noted within a low-lying cord. These are illustrated in (C). (D) T1 sagittal images showing proliferation of fat within the dural space, displacing and compressing the cord anteriorly.
Fig. 18 Intramedullary lipoma and dural lipomatosis (A) T2 sagittal image showing an intramedullary lipoma expanding the cord: Type VI. (B) T1 sagittal image showing an intramedullary lipoma noted within a low-lying cord. These are illustrated in (C). (D) T1 sagittal images showing proliferation of fat within the dural space, displacing and compressing the cord anteriorly.
 Fig. 19 Lipomyelomeningocele. (A) Myelogram, (B) T2 sagittal, and (C) T2 and (D) T1 axial images shows the herniation of the cord through the posterior spinal defect. A lipoma is noted in (D) with the lipoma placode interface located outside the spinal canal (arrowhead). This is a lipomyelomeningocele, as illustrated in (E).
Fig. 19 Lipomyelomeningocele. (A) Myelogram, (B) T2 sagittal, and (C) T2 and (D) T1 axial images shows the herniation of the cord through the posterior spinal defect. A lipoma is noted in (D) with the lipoma placode interface located outside the spinal canal (arrowhead). This is a lipomyelomeningocele, as illustrated in (E).
Type VI: Intramedullary lipoma ([Fig. 18]): integration of mesenchymal elements within the developing spinal cord results in the unusual intramedullary lipomas.[18]
Retained Medullary Cord
Defective apoptosis during late secondary neurulation results in a persistent medullary cord ([Fig. 21]). An absent filum terminale is characteristic of most RMCs.[19] They appear like a robust cord-like structure attached from the conus to the cul-de-sac. An altered shape of the conus is visible.
 Fig. 20 Spinal lipomas: a summary.
Fig. 20 Spinal lipomas: a summary.
 Fig. 21 Retained medullary cord. (A) T2 sagittal image showing a low-lying cord attached to a terminal lipoma by a thick, robust filum terminale. (B) T1 sagittal image showing a low-ending cord herniating into a CSF-filled sac. This is a cystomyelocele with possible RMC. (C) T2 sagittal image showing split cord malformation with a ventriculus terminalis cyst. (D) T2 sagittal image showing a low-lying cord with thick, robust filum terminale attaching to the cul-de-sac and altered conus shape.
Fig. 21 Retained medullary cord. (A) T2 sagittal image showing a low-lying cord attached to a terminal lipoma by a thick, robust filum terminale. (B) T1 sagittal image showing a low-ending cord herniating into a CSF-filled sac. This is a cystomyelocele with possible RMC. (C) T2 sagittal image showing split cord malformation with a ventriculus terminalis cyst. (D) T2 sagittal image showing a low-lying cord with thick, robust filum terminale attaching to the cul-de-sac and altered conus shape.
They are often misread as a low-lying conus or a tight filum terminale ([Table 7]). Although MRI signal intensity characteristics are the same as a low-lying conus making preoperative diagnosis difficult, intraoperative neuromonitoring helps distinguish between the two as an RMC is non-functional with no electrical activity. The symptoms are essentially due to associated cord tethering.
Table 7
RMCs and a close differential diagnosis: tight filum terminale
|
Retained Medullary Cord
|
Tight Filum Terminale
|
|
The shape of the conus altered/not demarcated
|
Conus shape unaffected
|
|
Low-lying conus
|
Low-lying/normal conus
|
|
Usually associated with tethered cord syndrome
|
Usually associated with tethered cord syndrome
|
|
Histological findings of RMCs include a dense glioneuronal core along with fibrofatty elements, an ependymal-lined central cavity, and vestigial nerve roots[19]
|
No evidence of glioneuronal elements on Histopathology
|
Table 8
Differences between a Limited dorsal myeloschisis and Congenital Dermal sinus
|
Limited dorsal myeloschisis (LDM)
|
Congenital dorsal dermal sinus (CDS)
|
|
Closed skin defect
|
Tract with lumen communicating with the skin opening
|
|
Fewer chances of infection and fewer neurological defects; hence, surgery needed only in cases of tethering
|
More chances of infection, more number of neurological defects, surgery even in asymptomatic cases in view of ascending infections
|
|
Tract: Intrathecal tracts clearly visible
|
Tract: Intrathecal tracts not visible
|
|
Attachment site: Clearly seen as hypointense round structure adhering to the dorsal aspect of the spinal cord above conus medullaris
|
Attachment site: variable and not clearly visible
|
|
Dorsal tenting of the spinal cord seen in nearly all patients
|
Dorsal tenting of the spinal cord is rare
|
|
Tethering is more common
|
Tethering is less common
|
|
Dermoid and (epi)dermoids are rare
|
Dermoid and (epi)dermoids are more common
|
Few variants ([Fig. 21]) may show an hourglass shape with the non-functional medullary cord located caudal to the conus with an intervening short filum terminale. A few cases of association with a subcutaneous meningocele or a dermal sinus have been known.[28] A few other cases showing an RMC associated with a lipoma are known.
Filar cysts: The etiology of filar cysts is unclear; however, it is postulated that it may be a pseudocyst-like structure formed by arachnoid reflections. Another theory says that could be a true cyst that is a different remnant of the terminal ventricle. Filar cysts are thought to be of no clinical significance and are a normal variant. They are located within the filum, in the midline, just distal to the conus medullaris. The difference in location helps to distinguish between the persistent terminal ventricle. Sometimes, these cysts are not well demonstrated on MRI.
Dermal Sinus
It is an epithelial-lined fistulous communication between the CNS or its meningeal covering and skin. It results from the focal incomplete disjunction between neuroectoderm and cutaneous ectoderm. Clinically, a midline dimple or ostium is found on the cutaneous surface and is commonly associated with cutaneous stigmata of underlying occult spinal dysraphism-like hairy nevus, hemangioma, or hyperpigmentation. The tract then ascends and opens into the spinal canal. Dermal sinus may be associated with intraspinal dermoid or epidermoid, which shows variable imaging findings depending on their contents. Dermoids usually appear hyperintense on both T1- and T2-weighted images, while epidermoids are hypointense on T1-weighted and hyperintense on T2-weighted images. CNS infection is a common complication because of fistulous communication and hence these cases require early surgical repair.
The dermal sinus is lined by squamous epithelium which is absent in the case of limited dorsal myeloschisis. Cord tenting is unusual at the site of attachment ([Fig. 23]).
 Fig. 22 Retained medullary cord (A) Thickened, abnormally low termination of cord without a clearly demarcated conus. (B) With apical lipoma. (C) With ventriculus terminalis cyst (D) Hour glass configuration of thickened conus. (E) Myelocystocele with herniation of the cord. (F) Partial herniation of the cord into the myelocystocele sac.
Fig. 22 Retained medullary cord (A) Thickened, abnormally low termination of cord without a clearly demarcated conus. (B) With apical lipoma. (C) With ventriculus terminalis cyst (D) Hour glass configuration of thickened conus. (E) Myelocystocele with herniation of the cord. (F) Partial herniation of the cord into the myelocystocele sac.
 Fig. 23 Dorsal dermal sinus. (A) Patient presented with a midline opening above the gluteal cleft. (B) T1 axial MinIP, (C) T2 sagittal images showing a hypointense tract extending from the skin opening into the spinal canal. (D) Different patient shows thickened T2 hypointense thickened tract with an ill-defined T2 hyperintensity within the spinal canal. (E) On contrast, there is thick peripheral enhancement. This is a dorsal dermal sinus with intraspinal abscess, due to infection.
Fig. 23 Dorsal dermal sinus. (A) Patient presented with a midline opening above the gluteal cleft. (B) T1 axial MinIP, (C) T2 sagittal images showing a hypointense tract extending from the skin opening into the spinal canal. (D) Different patient shows thickened T2 hypointense thickened tract with an ill-defined T2 hyperintensity within the spinal canal. (E) On contrast, there is thick peripheral enhancement. This is a dorsal dermal sinus with intraspinal abscess, due to infection.
Limited Dorsal Myeloschisis
Limited dorsal myeloschisis (LDM) is a closed form of spinal dysraphism, characterized by the presence of a fibroneural or fibrovasaculoneural tract connecting the skin lesion to the underlying spinal cord. It may be confused with congenital dermal sinus[20] ([Fig. 24]).
 Fig. 24 Flat LDM vs. dorsal dermal sinus. (A) Illustration shows an epithelium lined tract communicating from the skin to the spinal canal. An external opening is noted at the skin. Associated epidermoid cyst and complications such as intraspinal abscess are shown. (B) Illustration shows a flat LDM, where a fibroneural stalk attaches from the skin to the spinal cord, causing tenting of the cord at its junction.
Fig. 24 Flat LDM vs. dorsal dermal sinus. (A) Illustration shows an epithelium lined tract communicating from the skin to the spinal canal. An external opening is noted at the skin. Associated epidermoid cyst and complications such as intraspinal abscess are shown. (B) Illustration shows a flat LDM, where a fibroneural stalk attaches from the skin to the spinal cord, causing tenting of the cord at its junction.
Saccular LDMs ([Fig. 25]) occur due to accompanying hydromyelia of the fibroneural tract(swelling of the basal neural nodule with CSF or a dome-stalk type of saccular LDM). Nonsaccular “flat” LDMs may have cutaneous stigmata such as a pit, flat membrane, or a crater too. The fibroneural tract tethers the spinal cord ([Fig. 26]) causing characteristic tenting and the associated neurological symptoms.
 Fig. 25 Saccular LDM vs. cystomyelocele. (A) Illustration showing a thin fibroneural stalk attaching to the dome of a subcutaneous CSF-filled sac: dome stalk variant of saccular LDM. In (B), CSF fills and distends the canal of the fibroneural stalk, leading to a segmental myelocystocele variant of saccular LDM. (C) Illustration representing a myelocystocele, a different entity where the cord along with a dilated central canal herniates through a posterior defect.
Fig. 25 Saccular LDM vs. cystomyelocele. (A) Illustration showing a thin fibroneural stalk attaching to the dome of a subcutaneous CSF-filled sac: dome stalk variant of saccular LDM. In (B), CSF fills and distends the canal of the fibroneural stalk, leading to a segmental myelocystocele variant of saccular LDM. (C) Illustration representing a myelocystocele, a different entity where the cord along with a dilated central canal herniates through a posterior defect.
 Fig. 26 Limited dorsal myeloschisis. (A) T2 sagittal image shows a linear fibroneural track extending from a flat subcutaneous component to the cervical spinal canal, with tenting at the junction. (B) T2 sagittal image showing similar lesion in the lumbar region: these are flat (non-saccular) LDM. (C) T2 sagittal image showing a linear fibroneural track extending from the spinal canal to the dome of the dilated sac in the subcutaneous plane. This is the dome stalk variant of saccular LDM. (D, E) T2 sagittal images of a different patient show a sacular LDM with cystomyelocele, with the CSF tracking into the dilated central canal of the fibroneural track (black arrowheads) surrounded by a CSF filled sac (small white arrows). Tenting of the cord without extension of the cord itself is seen in (D) (large white arrows). (F) Figure shows a fibroneural tract passing anteriorly into a CSF-filled sac: possibly an anterior LDM.
Fig. 26 Limited dorsal myeloschisis. (A) T2 sagittal image shows a linear fibroneural track extending from a flat subcutaneous component to the cervical spinal canal, with tenting at the junction. (B) T2 sagittal image showing similar lesion in the lumbar region: these are flat (non-saccular) LDM. (C) T2 sagittal image showing a linear fibroneural track extending from the spinal canal to the dome of the dilated sac in the subcutaneous plane. This is the dome stalk variant of saccular LDM. (D, E) T2 sagittal images of a different patient show a sacular LDM with cystomyelocele, with the CSF tracking into the dilated central canal of the fibroneural track (black arrowheads) surrounded by a CSF filled sac (small white arrows). Tenting of the cord without extension of the cord itself is seen in (D) (large white arrows). (F) Figure shows a fibroneural tract passing anteriorly into a CSF-filled sac: possibly an anterior LDM.
A pearly white membranous crater is a characteristic cutaneous marker called ‘ Cigarette burn mark. Other common stigmata are dimple, pit, hypertrichosis, and hemangioma.
Low-lying cord: Persistent cord termination below L2–L3 after the first month of life in a full-gestation infant is abnormally low-lying. Axial T1-weighted images are most accurate in determining the conus level. The probability of tethered cord syndrome is more when a low-lying cord is associated with a tight filum terminale ([Fig. 27]).
 Fig. 27 Low-lying cord: the spectrum. (A) Normal cord termination with normal filum terminale. (B) Low-lying cord with a normal filum terminale. (C) Normal cord termination with a thick filum terminale. (D) Low-lying cord tight filum terminale.
Fig. 27 Low-lying cord: the spectrum. (A) Normal cord termination with normal filum terminale. (B) Low-lying cord with a normal filum terminale. (C) Normal cord termination with a thick filum terminale. (D) Low-lying cord tight filum terminale.
Tethered cord: It is a clinical syndrome usually associated with closed spinal dysraphism-like spinal lipomas ([Fig. 14]), the tight filum terminale ([Fig. 16 D]), split cord malformations ([Fig. 32]), caudal regression syndrome ([Fig. 35]), limited dorsal myeloschisis ([Fig. 25A]) meningoceles/meningomyeloceles ([Fig. 9]) due to anchoring of the spinal cord/neural placode or filum terminale to the surrounding tissues occurring primarily or secondary to scarring or formation of a dermoid post-surgery and not just due to a low-lying cord or a thick filum terminale. The tethering causes functional neurological deterioration resulting in motor and sensory dysfunction, muscle atrophy, urinary incontinence, decreased or hyperactive reflexes, spastic gait, and other deformities.
Persistent fifth ventricle: Evidence of a fifth ventricle not accompanied by other pathologies is a frequent finding that does not have pathological significance during the first 5 years of life.[21] It is located immediately above the filum terminale and without contrast enhancement, differentiating it from other cystic lesions of the conus medullaris ([Fig. 22C]).
Disorders of Midline Notochordal Integration
The process of fusion of paired notochordal anlagen to form a single midline notochordal process is called midline notochordal integration. Any abnormality at this stage results in longitudinal cord splitting. The most important conditions in this group are neurenteric cysts, dorsal enteric fistula ([Fig. 28]), and diastematomyelia.
 Fig. 28 Spectrum of neuroenteric remnants. (A) Dorsal enteric diverticulum. (B) Dorsal enteric fistula (C) Postvertebral neuroenteric cyst. (D) Intraspinal neuroenteric cyst. (E) Prevertebral neuroenteric cyst. (F) Dorsal enteric sinus.
Fig. 28 Spectrum of neuroenteric remnants. (A) Dorsal enteric diverticulum. (B) Dorsal enteric fistula (C) Postvertebral neuroenteric cyst. (D) Intraspinal neuroenteric cyst. (E) Prevertebral neuroenteric cyst. (F) Dorsal enteric sinus.
Dorsal enteric fistula and neurenteric cyst: The most severe form of disorder of midline notochordal integration is a dorsal enteric fistula, which is a fistulous communication between dorsal skin surface and bowel.
The spectrum of dorsal enteric remnants consists of (1) dorsal enteric fistula, (2) dorsal enteric sinus, (3) dorsal enteric cyst (neurenteric cyst), and (4) dorsal enteric diverticulum.
The dorsal enteric fistula ([Fig. 29]) is due to a persistent connection between the endoderm and ectoderm, resulting in the splitting of the notochord. The fistula traverses the prevertebral soft tissue, the vertebral bodies, and the spinal canal with its contents. Any portion of this tract may involute or become fibrous, leading to a fistula or a cyst.
 Fig. 29 Dorsoenteric fistula. (A) Patient presented with a large swelling over the back. (B) Ultrasonogram showing multiple dilated bowel loops within the swelling. (C) CT VRT image showing the split vertebral column with duplication of the vertebral bodies with posterior vertebral defect. (D) Myelogram, (E) T2 sagittal, (F) T2 coronal images showing herniation of bowel loops through the split cord. (G) Intraoperative image showing multiple herniated loops.
Fig. 29 Dorsoenteric fistula. (A) Patient presented with a large swelling over the back. (B) Ultrasonogram showing multiple dilated bowel loops within the swelling. (C) CT VRT image showing the split vertebral column with duplication of the vertebral bodies with posterior vertebral defect. (D) Myelogram, (E) T2 sagittal, (F) T2 coronal images showing herniation of bowel loops through the split cord. (G) Intraoperative image showing multiple herniated loops.
A Neurenteric cyst ([Fig. 30]) is a localized form of dorsal enteric fistula. These cysts are typically seen in the extramedullary intradural compartment of the cervicothoracic spine; however, may be seen in other locations such as subcutaneous, intramedullary, or prevertebral planes. On MRI, neurenteric cysts usually appear iso-to-hyperintense to CSF on both T1-and T2-weighted images due to high protein content and show absent contrast enhancement.[22]
 Fig. 30 Neuroenteric cyst. (A) T1 coronal image and (B) T2 axial image showing neuroenteric cyst appearing iso-to-hyperintense to CSF on both T1- and T2-weighted images. (C, D) T2 axial images showing an anterior mediastinal cystic lesion, communicating with the spinal canal through a vertebral cleft (arrowhead).
Fig. 30 Neuroenteric cyst. (A) T1 coronal image and (B) T2 axial image showing neuroenteric cyst appearing iso-to-hyperintense to CSF on both T1- and T2-weighted images. (C, D) T2 axial images showing an anterior mediastinal cystic lesion, communicating with the spinal canal through a vertebral cleft (arrowhead).
An isolated (epi)dermoid appearing T2 hyperintense is an unusual differential in the case of intraspinal neurenteric cysts. The dermoid/epidermoid cyst occurs due to epithelial rests within the neural tube ([Fig. 31]).
 Fig. 31 Intraspinal dysraphism: epidermoid. (A) T2 sagittal image showing a heterogenous, T2 hyperintense mass lesion within the dural space compressing the cord anteriorly. This is an epidermoid cyst. Dermoids and epidermoids are differential diagnoses and to be considered for a neurenteric cyst.
Fig. 31 Intraspinal dysraphism: epidermoid. (A) T2 sagittal image showing a heterogenous, T2 hyperintense mass lesion within the dural space compressing the cord anteriorly. This is an epidermoid cyst. Dermoids and epidermoids are differential diagnoses and to be considered for a neurenteric cyst.
The dorsal enteric sinus, a remnant of the posterior portion of the tract, has an opening on the skin surface. Dorsal enteric cysts are trapped remnants of the middle portion of the tract, found in the intraspinal or paraspinal compartments.
The dorsal enteric diverticulum is a tubular diverticulum arising from the bowel and represents a remnant of the anterior portion of the tract
There exists a high incidence of associated urogenital malformations and anorectal malformations.
Diastematomyelia
It is the most common form of defective midline notochordal integration. Due to defective midline integration, there are two notochordal processes each of which induces the formation of separate neural plates with primitive streak tissue in between them. The development of the primitive streak tissue decides the type of diastematomyelia.
Pang et al. coined a new term called split cord malformations and divided it into two types. In type I diastematomyelia, the intervening primitive streak develops into a bone or cartilage, resulting in two hemicords in different dural sacs separated by an osseocartilaginous septum. Each of them includes a central canal, one dorsal, and one ventral root ([Fig. 32]). Type 1 is more common than type II. In type II diastematomyelia, the primitive streak is reabsorbed or forms a fibrous septum with the hemicords lying within the same dural sac. Diastematomyelia is commonly associated with vertebral anomalies and hydromyelia. A high-lying hairy tuft over a child's back is a reliable indicator for underlying diastematomyelia.
 Fig. 32 Diastematomyelia. (A) Image shows hypertrichosis (fawn's tail) over the lumbar region. (B) T2 axial image showing diastematomyelia with a common dural sac: type 1. (C) T2 axial image showing diastematomyelia with a bony bar separating the hemicords: type 2. (D) T2 axial image showing type 1 diastematomyelia at the cervical level, a rare entity. (E) T2 axial image showing the cord cleft without splitting of the hemicords. (F) T1 coronal and (G) CT images showing duplication of the vertebral column. (H) There is a lipoma attached to the left hemicord, with the herniation of the lipoma and hemicord posteriorly through the spinal defect.
Fig. 32 Diastematomyelia. (A) Image shows hypertrichosis (fawn's tail) over the lumbar region. (B) T2 axial image showing diastematomyelia with a common dural sac: type 1. (C) T2 axial image showing diastematomyelia with a bony bar separating the hemicords: type 2. (D) T2 axial image showing type 1 diastematomyelia at the cervical level, a rare entity. (E) T2 axial image showing the cord cleft without splitting of the hemicords. (F) T1 coronal and (G) CT images showing duplication of the vertebral column. (H) There is a lipoma attached to the left hemicord, with the herniation of the lipoma and hemicord posteriorly through the spinal defect.
Diastematomyelia can be classified into three types depending on location 1) lumbar (most common), 2) lumbosacral, 3) thoracolumbar, and 4) cervicothoracic.
Composite types of split cord malformations (multi-level split cord malformations) are those with multiple levels of split cord in the same patient. Another classification, depending on the length of involvement, is divided into short-segment and long-segment types. When the split cord malformation is partial, it is called partial diastematomyelia (spinal cord cleft), seen associated with Klippel–Feil syndrome.[23] Split cord malformations include a wide spectrum of spinal abnormalities, which may range from simple diastematomyelia to complete duplication of the spinal column with two hemicords.
Vertebral Duplication
Vertebral duplication of split cord malformation (SCM) is a rare abnormality. Affected individuals present with clinical pictures ranging from mild-to-very severe symptoms which may be associated with gastrointestinal abnormalities such as neurenteric fistula, omphalocele or neurologic abnormalities such as myelomeningocele. Dorsolumbar and lumbar regions are the most affected sites. Vertebral duplication as such has double cords which are not separated by a septum unlike in Type 1 diastematomyelia but may be associated with diastematomyelia at a different level ([Fig. 32F], G). Each cord had its own spinal canal within its own set of vertebrae.
Segmental spinal dysgenesis: Segmental spinal dysgenesis is characterized by a localized deformity of the thoracolumbar, lumbar, lumbosacral spine, which is associated with localized agenesis/dysgenesis spine and spinal cord. This occurs due to an abnormality of an intermediate segment of notochord occurring during gastrulation. SSD is a rare, complex, congenital, closed spinal dysraphism that meets the following criteria[26]: (1) presence of congenital paraparesis/paraplegia with lower limb abnormalities; (2) multiple (more than one) formation and segmentation anomalies of the vertebra with or without kyphoscoliosis; (3) absent or malformed segment of the spinal cord and underlying nerve roots involving any spinal segment from the cervical to sacral region; and (4) visualization of the segment of spinal cord distal to the interrupted cord ([Fig. 33]).
 Fig. 33 Segmental spinal dysgenesis. T2 sagittal image showing focal dysgenesis of the spinal cord, with a rounded termination of the upper segment (arrowhead) and an abnormal sacral segment (arrow)
Fig. 33 Segmental spinal dysgenesis. T2 sagittal image showing focal dysgenesis of the spinal cord, with a rounded termination of the upper segment (arrowhead) and an abnormal sacral segment (arrow)
It usually requires a multidisciplinary management approach based not only on the extent and severity of the dysgenetic cord but also on the cause (type 1 primary cord hypoplasia or type 2 secondary cord hypoplasia due to dysmorphic vertebra) of the spinal dysgenesis for further surgical management.[26]
Caudal agenesis/caudal regression syndrome: It is characterized by partial or total agenesis of the spinal column and is commonly associated with genital anomalies, anal imperforation, pulmonary hypoplasia, renal aplasia or dysplasia, and limb abnormalities. This may be associated with syndromic complexes such as OEIS, VACTERL, and the Currarino triad (partial sacral agenesis, anorectal malformation, and presacral mass), wherein aberrations from the stage of midline notochordal integration and mesodermal proliferation result in combined abnormalities ([Fig. 34]).[9]
 Fig. 34 Anterior lipomyelocele and an absent right hemisacrum. (A) T2 axial and (B) T1 sagittal images showing a lipoma with the lipoma: placode interface lying within the spinal canal. The lipoma herniates anteriorly and to the right through an absent right hemisacrum. The triad of sacral defect, anorectal malformation, and anterior spinal mass is Currarino's triad.
Fig. 34 Anterior lipomyelocele and an absent right hemisacrum. (A) T2 axial and (B) T1 sagittal images showing a lipoma with the lipoma: placode interface lying within the spinal canal. The lipoma herniates anteriorly and to the right through an absent right hemisacrum. The triad of sacral defect, anorectal malformation, and anterior spinal mass is Currarino's triad.
A low crown–rump length may be the first sign of CRS in the first trimester. At later stages, there is the abrupt interruption of the spine at dorsal, lumbar, or sacral level with femurs fixed in a “V” pattern.
Caudal regression syndrome is broadly divided into two types. In type I caudal regression syndrome, both caudal cell mass and notochord formation are affected, resulting in the high position (D12 vertebra) and abnormal termination of conus medullaris. There is an association of vertebral aplasia in the majority of patients.
In type II caudal regression syndrome, there is abnormal development of the caudal cell mass with unaffected true notochord formation. Hence, there is defective secondary neurulation with normal primary neurulation. As a result, only the most caudal part of conus medullaris is absent in type II CA ([Fig. 35]). Vertebral dysgenesis is less severe in these cases and these patients present with tethered cord syndrome as the conus in these cases is stretched and tethered. In the case of lumbosacral classification, Pang's classification (1993) describes five types:
Type 1:Total sacral agenesis (some lumbar vertebrae are missing)
Type II: Total sacral agenesis (lumbar vertebrae are not involved)
Type III: Subtotal sacral agenesis, S1 present
Type IV: Hemisacrum
Type V: Coccygeal agenesis[24]
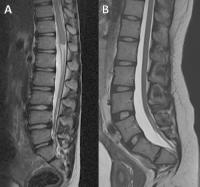 Fig. 35 Caudal regression syndrome. (A) T2 sagittal image showing the high position of a bulbous conus medullaris with abnormal termination and partial sacral agenesis. This is Type 1 caudal regression syndrome. (B) T2 sagittal image showing a low-lying cord tethered by a thick filum terminale, with partial sacral agenesis. This is type 2 caudal regression syndrome.
Fig. 35 Caudal regression syndrome. (A) T2 sagittal image showing the high position of a bulbous conus medullaris with abnormal termination and partial sacral agenesis. This is Type 1 caudal regression syndrome. (B) T2 sagittal image showing a low-lying cord tethered by a thick filum terminale, with partial sacral agenesis. This is type 2 caudal regression syndrome.
Sacrococcygeal Teratoma
Although sacrococcygeal teratoma is a tumor, the embryological origin is from the pluripotent cells of caudal cell mass. This tumor usually presents as a large complex solid–cystic mass caudal to the coccyx. Most of them are benign and contain derivatives from all three germ layers. Based on the presence of external and internal components, they are divided into four types: Type 1: primarily external, type 2: Equal external and internal portions, Type 3: primarily internal, and Type 4: entirely internal.
This could be diagnosed in antenatal ultrasound. In MRI, the mass has variable appearance depending on the internal contents, namely, fat, soft tissue, fluid, or calcium with a heterogeneous enhancement of solid components of the mass on post-contrast study.
The migration of pleuripotent cells to unusual locations such as the level of cervical, dorsal, or lumbosacral vertebrae may also result in teratomas.[28]
Spinal/Sacral Chordoma
Spinal/sacral chordomas are aggressive rare malignant masses of notochordal origin presenting as a firm mass.[29] They occur most commonly in the sacrococcygeal region (30–35%). They present as a large destructive T1/T2 heterointense lesion with SWI/GE showing variable intralesional hemorrhage, suggested by the presence of a blooming artifact.






