

Subscribe to RSS
DOI: 10.1055/s-0041-1740944
Enantioselective aza-Michael Cyclization Reaction Catalyzed by Quinine-Derived Monoquaternary Ammonium Salts: an Effective Route to Synthesize Letermovir
Authors


Abstract
A series of mono- or bis-quaternary ammonium salts derived from cinchonidine or quinine was synthesized and screened as potent phase-transfer catalysts for the reaction of aza-Michael cyclization, the key step in the synthesis of letermovir. During the reaction of aza-Michael cyclization, the screened monoquaternary ammonium salt quinine derivative Q1 transferred 7 to 8 with 91.9% yield and 58% ee. The application of Q1 was preferred, due to its enantioselectivity, the possibility of reuse, and the lower cost in large-scale preparation. Furthermore, the racemization condition of letermovir enantiomer was also explored for the possibility to develop the resolution/racemization process. With the optimal catalyst Q1 in hand, the synthesis of letermovir may be more convenient and economical in the future.
Keywords
letermovir - monoquaternary ammonium salts - phase-transfer catalyst - enantioselective - aza-Michael cyclization - racemizationIntroduction
Human cytomegalovirus (HCMV) infections remain a prevalent cause for morbidity and mortality in immunosuppressed solid-organ and bone-marrow transplant patients, and the most frequent cause of opportunistic viral infection following transplantation.[1] [2] The incidence of HCMV infection in the general population ranges from 36 to 90%, and the seropositivity increases with age and tends to be highest among lower socioeconomic groups.[3] [4] Letermovir is a novel antiviral compound currently used for the prevention of HCMV infections. Before the approval of letermovir, the first line of systemic anti-HCMV drugs includes ganciclovir, foscarnet, and cidofovir, which are derivatives of nucleosides and all target the DNA polymerase, yet, associated with significant side effects such as clinically significant myelosuppression,[5] [6] leading to the limited use of them in HCMV therapy.[7] Thus, developing a safe and effective agent for HCMV therapy has been an area of unmet need that is urgently needed in cytomegalovirus (CMV) management. In November 2017, letermovir was approved by the U.S. Food and Drug Administration for CMV prophylaxis in adult allogeneic CMV-seropositive hematopoietic stem cell transplant recipients.[8] As a novel 3,4-dihydro-quinazoline-4-yl-acetic acid derivative, letermovir represents a new class of nonnucleoside CMV inhibitors. Unlike other anti-CMV therapies, letermovir disturbs the late stages of viral replication rather than inhibiting the viral DNA polymerase,[9] [10] with no significant myelotoxicity or nephrotoxicity appearing.[11]
The commercial synthetic route to letermovir (1) was reported by Merck and Co., Inc.[12] Letermovir was obtained from 2 through eight steps, including heck reaction, urea condensation, dehydration, electrophilic substitution, asymmetrical aza-Michael cyclization, chiral resolution, and hydrolysis. The key step is the enantioselective aza-Michael cyclization with bis-quaternary ammonium salt cinchonidine derivative A using as the phase-transfer catalyst (PTC) ([Scheme 1]). PTC A is not commercially available and needs to be prepared from a very expensive material, cinchonidine. Moreover, as a di-benzyl-substituted bis-quaternary ammonium salt, PCT A is hard to be recycled due to its instability in the reaction and the work-up process. This made us find a better PTC for the replacement.

Inspired by our previous work on the synthesis of dezocine, 17 monoquaternary ammonium salts, derived from cinchonidine or quinine ([Scheme 2]), were screened as the PTCs for the asymmetrical alkylation of 1-methyl-7-methoxy-2-tetralone and some of them were found to function well.[13] As a continuation, it is interesting to evaluate their effect on the asymmetrical aza-Michael cyclization. Herein, we reported an effective and economical route to synthesize letermovir using a quinine-derived monoquaternary ammonium salt (Q1) as the PTC in the reaction of aza-Michael cyclization. When we used Q1 as the catalyst, the cost of the catalyst was far cheaper in comparison to that of PCT A; furthermore, the catalyst can be reused and the ee value of the target product (8) was 58%. Thus we consider that Q1 was a better catalyst for the aza-Michael cyclization.

Materials and Methods
All solvents and reagents were purchased from commercial sources and were used without further purification except compound 2, which was used after distillation. 2 (2-bromo-6-fluorobenzenamine) was purchased from Fuxin Jintelai Fluorin Chemical Co., Ltd. Quinine and cinchonidine were purchased from Shanghai Macklin Biochemical Co., Ltd. (+)-di-p-Toluoyl-D-tartaric acid (D-(+)-DTTA) was purchased from Shanghai Haoyuan Chemexpress Co., Ltd. Melting points were determined on a Büchi Melting Point M-565 apparatus. 1H spectra were recorded using a Bruker 400 MHz spectrometer with chlorotrimethylsilane as an internal standard. Mass spectra were recorded with a quadrupole time-of-flight mass spectrometer using electrospray positive ionization (ESI + ). The enantiomeric ratio was determined by high-performance liquid chromatography (HPLC, Agilent 1260) with a chiral column (CHIRALPAK IA). The eluent was the solution of hexane/isopropyl alcohol (75:25) with 0.1% diethylamine being added, and the detection wavelength was 254 nm. Specific rotations were determined on a Rudolph Research Analytical automatic polarimeter IV. All reactions were monitored by thin layer chromatography, which were performed on silica gel GF254. Column chromatography was performed on silica gel (200–300 mesh) purchased from Qingdao Ocean Chemical Company of China.
General Procedure for the Preparation of Methyl-(S)-2-(8-fluoro-3-(2-methoxy-5-(trifluoromethyl)phenyl)-2-(4-(3-methoxyphenyl)-piperazin-1-yl)-3,4-dihydroquinazolin-4-yl)acetate(2S,3S)-2,3-bis((4-methylbenzoyl)oxy)succinate ethyl acetate solvate (9)
To a stirred mixture of toluene (700 mL) and 7 (70 g, 96.68 mmol) was added K3PO4 aqueous solution (1 mol/L, 145 mL) under an ice bath. After 20 minutes, the mixture became clarified. The organic layer was separated, and K3PO4 aqueous solution (0.43 mol/L, 272 mL) was added at 0 to 5°C followed by the addition of Q1 (3.05 g, 4.83 mmol). After warming to room temperature, the reaction solution was stirred for 5 hours until 7 was consumed, and then glycolic acid solution (1 mol/L, 145 mL) was added. The resulting mixture was stirred for 10 minutes at 45°C, and then cooled down. The organic layer was separated and evaporated to give 8 as yellow oil (52.1 g, yield: 91.9%; HPLC chiral purity: 8a:8b/78:22).
The oil 8 was dissolved in ethyl acetate (1,000 mL), and then D-(+)-DTTA (34.34 g, 88.9 mmol) was added. The mixture was stirred overnight and filtered. The filtrate was washed with ethyl acetate (100 mL) and dried at 75°C to afford 9 (66.83 g, yield: 77.3%). Mp 128–129°C. 1H NMR (400 MHz, DMSO-d 6 + D2O) δ 7.91–7.89 (d, J = 8.0 Hz, 4H), 7.58–7.55 (dd, J1 = 12.0 Hz, J2 = 4.0 Hz, 1H), 7.41–7.39 (d, J = 8.0 Hz, 4H), 7.28–7.26 (d, J = 8.0 Hz, 1H), 7.07 (t, J = 12.0 Hz, 2H), 6.93–6.87 (m, 2H), 6.47–6.44 (dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 1H), 6.40–6.36 (m, 2H), 5.82 (s, 2H), 4.94 (t, J = 8.0 Hz, 1H), 4.02 (q, J = 12.0 Hz, 2H), 3.87 (brs, 3H), 3.69 (s, 3H), 3.64 (s, 3H), 3.47–3.37 (m, 5H), 2.96–2.94 (m, 2H), 2.85–2.81 (m, 2H), 2.57 (m, 1H), 2.41 (s, 6H), 1.99 (s, 2H), 1.24 (s, 1H), 1.18 (t, J = 8.0 Hz, 2H). MS (ESI + ) m/z calcd. for C30H30F4N4O4 [M + H]+ 587.22, found 587.21.
General Procedure for the Preparation of (S)-2-(8-Fluoro-3-(2-methoxy-5-(trifluoromethyl)-phenyl)-2-(4-(3-methoxyphenyl)piperazin-1-yl)-3,4-dihydroquinazolin-4-yl)acetic acid (Letermovir, 1)
DTTA salt 9 (40.0 g, 40.12 mmol) was slurried in ethyl acetate (400 mL), and then Na2HPO4 aqueous solution (0.5 mol/L, 206 mL) was added. The mixture was stirred for 0.5 hours. The separated organic phase was concentrated to get a white bubble-shaped solid, which was dissolved in MeOH (240 mL), and in NaOH aqueous solution (1 mol/L, 120 mL). The mixture was stirred at room temperature for 1.5 hours, acidified with HCl (3 mol/L, 30 mL) to pH value of 6 to 7, and then extracted with ethyl acetate (200 mL × 3). The combined organic phase was washed with brine, dried with MgSO4, and concentrated to afford letermovir (22.6 g; yield: 96%; HPLC achiral purity: 99.5%, and chiral purity: 99.4%). Mp 117–119°C. 1H NMR (400 MHz, CDCl3 + D2O) δ 7.51–7.49 (d, J = 8.0 Hz, 1H), 7.28 (s, 1H), 7.13 (t, J = 8.0 Hz, 1H), 7.03–6.96 (m, 3H), 6.88–6.86 (d, J = 8.0 Hz, 1H), 6.44–6.38 (m, 2H), 6.32 (t, J = 4.0 Hz, 1H), 4.99 (t, J = 8.0 Hz, 1H), 3.76 (s, 6H), 3.56 (s, 4H), 2.96–2.95 (m, 4H), 2.88 (s, 1H), 2.72 (dd, J1 = 16.0 Hz, J2 = 8.0 Hz, 1H). MS (ESI + ) m/z calcd. for C29H28F4N4O4 [M + H]+ 573.20, found 573.24.
Results and Discussion
PTC Screening
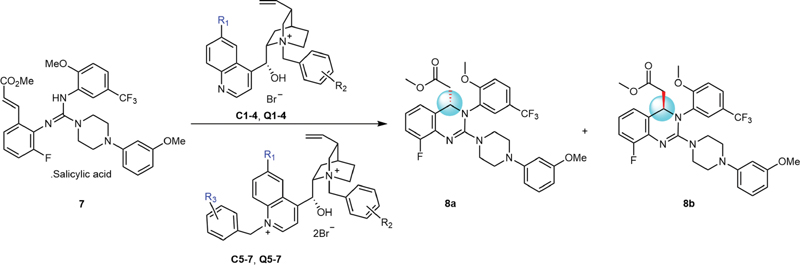
The literature suggested that alkaloids bearing benzyl with electron-withdrawing groups are beneficial for the enantioselective aza-Michael addition.[12] Therefore, eight monobenzyl quaternary ammonium salts from cinchonidine (C1–C4) and quinine (Q1–Q4) were adopted to evaluate the structure–enantioselectivity relationship, and six bis-quaternary ammonium salts (C5–C7 and Q5–Q7) were used for comparison.
The aza-Michael cyclization of compound 7 was performed in toluene/H2O with the base of K3PO4 and the catalysis of PTC. When compound 7 was completely consumed, the target product was purified by flash chromatography followed by enantioselectivity analysis through a chiral HPLC determination. The results are listed in [Table 1].
|
|
||||||
|---|---|---|---|---|---|---|
|
Entry |
Catalyst |
R1 |
R2 |
R3 |
Yield[a] |
8a:8b[b] |
|
1 |
C1 |
H |
3,5-CF3 |
– |
80.0% |
79:21 |
|
2 |
C2 |
H |
3-CF3 |
– |
92.0% |
71:29 |
|
3 |
C3 |
H |
4-CF3 |
– |
76.0% |
65:35 |
|
4 |
C4 |
H |
3,4-Cl |
– |
68.3% |
71:29 |
|
5 |
C5 |
H |
3,5-CF3 |
3-CF3 |
77.0% |
73:27 |
|
6 |
C6 |
H |
3,5-CF3 |
3,5-CF3 |
81.5% |
64:36 |
|
7 |
C7 |
H |
3-CF3 |
3-CF3 |
66.7% |
59:41 |
|
8 |
Q1 |
–OCH3 |
3,5-CF3 |
– |
91.9% |
79:21 |
|
9 |
Q2 |
–OCH3 |
3-CF3 |
– |
84.9% |
69:31 |
|
10 |
Q3 |
–OCH3 |
4-CF3 |
– |
91.1% |
65:35 |
|
11 |
Q4 |
–OCH3 |
3,4-Cl |
– |
96.2% |
69:31 |
|
12 |
Q5 |
–OCH3 |
3,5-CF3 |
3-CF3 |
67.5% |
71:29 |
|
13 |
Q6 |
–OCH3 |
3,5-CF3 |
3,5-CF3 |
63.3% |
63:37 |
|
14 |
Q7 |
–OCH3 |
3-CF3 |
3-CF3 |
74.9% |
62:28 |
Abbreviation: PTCs, phase-transfer catalysts.
a Isolated as a mixture of 8a and 8b.
b Enantioselectivity was determined by chiral HPLC analysis.
Our data suggested that monoquaternary ammonium salts, both from cinchonidine (C1–C4) and quinine (Q1–Q4), had a good effect on enantioselectivity; for example, both entry 1 and entry 8 gave the enantiomeric ratio of 8a:8b at 79:21. This result was meaningful, because the commercial price of quinine is about one-20th of that of cinchonidine. Besides, the results further demonstrated that the benzyl substituted with two trifluoromethyl was better than that substituted with one trifluoromethyl (C1 versus C2 or C3; Q1 versus Q2 or Q3), so compound Q1 could be considered as the potential PTC. The screening results also suggested that other electron-withdrawing groups in benzyl such as 3,4-dichloro did not improve the enantiomeric ratio.
Evidence suggested that the bisbenzyl quaternary ammonium salt C5, a cinchonidine derivative, was reported to have good enantioselectivity, which was further confirmed in entry 5 (8a:8b = 73:27).[12] However, other bis-quaternary ammonium salts C6, C7, Q6, and Q7 gave the worse enantiomeric ratio of the reaction in comparison to that of Q5. Furthermore, we found that the bi-quaternary ammonium salts were hard to be recovered, and the preparation of them was also complex. Bis-quaternary PTCs, if applied in large scale, would cost much more than the monoquaternary ones. Thus, Q1 was selected for the following large-scale reaction.
Proposed Catalytic Mechanism
According to the result of the PTC screening, the catalytic mechanism was proposed as shown in [Scheme 3]. Briefly, an anion on the nitrogen of the guanidine moiety of 7 was formed through base treatment, then, the anion intermediate interacted with the catalyst to form the transition state through an ionic bond in the interface of the bi-phase (toluene/water), as well as a hydrogen bond between the carbonyl oxygen atom of the substrate and the hydroxyl of the catalyst. Meanwhile, the phenyl on the substrate provided the π–π interaction with the benzyl of the catalyst. Under the action of the catalyst, N- selectively attacked the substrate in the opposite direction to the π–π interaction site to complete the stereoselective aza-Michael cyclization.

Optimized Large-Scale Synthesis of Letermovir (1)
The optimal PTC Q1 encouraged us to optimize the total synthetic route of letermovir that was reported by Merck and Co., Inc. [Scheme 4] illustrates an optimized process of letermovir (1) synthesis in comparison to a reported study.[12] Briefly, compound 7 (as a salicylic acid salt) was obtained from 2-bromo-6-fluoroaniline through five steps and underwent the stereoselective aza-Michael cyclization under the catalysis of Q1. After DTTA resolution, 9 was hydrolyzed in MeOH/H2O to give the target 1.

However, some trouble was encountered in practice. During dissociation of the freebase from 7 in the presence of base K3PO4 at ca. 30°C, some percentage (10–30%) of the background aza-Michael cyclization already taken place before the addition of PTC. It was pleased to find that if this dissociation was performed at 0 to 5°C, the background reaction could be eliminated. In the presence of Q1 and K3PO4, 8 was obtained from 7 in a yield of 68.3% with 58% ee. After the salt formation with D-(+)-DTTA in ethyl acetate, the ee value increased to 99.3%. The catalyst was recovered by column chromatography from the mother liquor and could be reused in the same reaction without the loss of yield or ee value.
In the hydrolysis step, a long reaction time (at least 20 hours) was required when the hydrolysis was performed in NaOH aqueous solution at 60°C, according to a reported study,[12] which increased the risk of the racemization of the product. To our delight, when the reaction solution was replaced with NaOH aqueous solution/MeOH (v/v = 1:2), the reaction could be performed at room temperature and the reaction time could be shortened to approximately 1 hour, giving 1 in a yield of 95%, an achiral purity of 99.5%, and a chiral purity of 99.4%.
A small amount of racemization was also observed in the hydrolysis step, especially if the reaction lasted for a long time. To verify the possibility of the racemization of the product and develop the process to reuse the enantiomer of letermovir, experiments on the racemization of the enantiomer were further designed. The mother liquor (8a:8b/9.3:90.7) from D-(+)-DTTA salt formation was recovered and hydrolyzed into the acid, which was used for the racemization, and the result is included in [Table 2]. Our data suggested that sodium methoxide in acetonitrile or methanol under reflux did not realize the racemization. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was not the ideal base. Finally, racemization could be performed under the condition of 2 equiv. of KOH in DMSO at 100°C (entry 5) with R:S = 52.7: 47.2 identified by chiral HPLC. This result laid the foundation to develop a recycling process of resolution/racemization to reuse the enantiomer of letermovir.
|
Entry |
Base |
Solvent |
Temperature (°C) |
Time (h) |
Specific rotation |
Result |
|---|---|---|---|---|---|---|
|
1 |
Sodium methoxide[a], 2 equiv. |
Acetonitrile |
Reflux |
72 |
221.2 (c = 1, MeOH)[b] |
No racemization |
|
2 |
Sodium methoxide[a], 2 equiv. |
Methanol |
Reflux |
48 |
235.2 (c = 1, MeOH)[b] |
No racemization |
|
3 |
DBU, 2 equiv. |
Acetonitrile |
Reflux |
72 |
237.3 (c = 1, MeOH) [b] |
No racemization |
|
4 |
DBU, 3 equiv. |
DMSO |
65 |
16 |
268 (c = 1, MeOH) [b] |
No racemization |
|
5 |
KOH, 2 equiv. |
DMSO |
100 |
10 |
6.5[c] |
Racemization |
|
6 |
KOH, 4 equiv. |
DMSO |
100 |
10 |
– |
Several impurities |
a 30% solution in methanol.
b c = 1 means 1 g/100mL.
c R:S = 52.7: 47.2, determined by chiral HPLC analysis.
Conclusion
In summary, an optimal catalyst Q1 (quinine-derived monoquaternary ammonium salt) was explored, due to its good enantioselectivity, easy preparation from cheap material, as well as the possibility of reuse. With the use of the selected Q1, the synthesis of letermovir would be more convenient and economical. Furthermore, the hydrolysis (the last step) of the total synthesis route of letermovir was also improved to ensure the purity of the target product. The racemization condition of the enantiomer of letermovir was explored for the possibility to develop the resolution/racemization process so that the enantiomer could be completely used.
Conflict of Interest
None.
Supplementary Information
Detailed information for the synthesis of the catalysts C1–C8, Q1–Q7, as well as the chiral HPLC diagrams of 8 is provided and available online only.
-
References
- 1 Schubert L, Fisecker L, Thalhammer F, Burgmann H, Steininger C. Letermovir for the compassionate therapeutic use of cytomegalovirus infection. Eur J Clin Microbiol Infect Dis 2021; 40 (02) 435-439
- 2 Suárez NM, Blyth E, Li K. et al. Whole-genome approach to assessing human cytomegalovirus dynamics in transplant patients undergoing antiviral therapy. Front Cell Infect Microbiol 2020; 10: 267
- 3 Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis 2010; 50 (11) 1439-1447
- 4 Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis 2006; 43 (09) 1143-1151
- 5 Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23 (04) 689-712
- 6 El Helou G, Razonable RR. Safety considerations with current and emerging antiviral therapies for cytomegalovirus infection in transplantation. Expert Opin Drug Saf 2019; 18 (11) 1017-1030
- 7 Othumpangat S, Noti JD, Ray SD. Chapter 25 - Antiviral drugs. In: Ray SD, ed. Side Effects of Drugs Annual. Amsterdam: Elsevier; 2017. (39): 269-282
- 8 Center For Drug Evaluation And Research. 209939Orig1s000; 209940Orig1s000. 2017
- 9 Lischka P, Hewlett G, Wunberg T. et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 2010; 54 (03) 1290-1297
- 10 Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 2011; 85 (20) 10884-10893
- 11 Imlay HN, Kaul DR. Letermovir and maribavir for the treatment and prevention of cytomegalovirus infection in solid organ and stem cell transplant recipients. Clin Infect Dis 2021; 73 (01) 156-160
- 12 Humphrey GR, Dalby SM, Andreani T. et al. Asymmetric synthesis of letermovir using a novel phase-transfer-catalyzed aza-michael reaction. Org Process Res Dev 2016; 20 (06) 1097-1103
- 13 Li R, Liu Z, Chen L, Pan J, Zhou W. Enantioselective phase-transfer catalyzed alkylation of 1-methyl-7-methoxy-2-tetralone: an effective route to dezocine. Beilstein J Org Chem 2018; 14: 1421-1427
Address for correspondence
Publication History
Received: 20 October 2021
Accepted: 11 November 2021
Article published online:
30 December 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Schubert L, Fisecker L, Thalhammer F, Burgmann H, Steininger C. Letermovir for the compassionate therapeutic use of cytomegalovirus infection. Eur J Clin Microbiol Infect Dis 2021; 40 (02) 435-439
- 2 Suárez NM, Blyth E, Li K. et al. Whole-genome approach to assessing human cytomegalovirus dynamics in transplant patients undergoing antiviral therapy. Front Cell Infect Microbiol 2020; 10: 267
- 3 Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin Infect Dis 2010; 50 (11) 1439-1447
- 4 Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis 2006; 43 (09) 1143-1151
- 5 Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 2010; 23 (04) 689-712
- 6 El Helou G, Razonable RR. Safety considerations with current and emerging antiviral therapies for cytomegalovirus infection in transplantation. Expert Opin Drug Saf 2019; 18 (11) 1017-1030
- 7 Othumpangat S, Noti JD, Ray SD. Chapter 25 - Antiviral drugs. In: Ray SD, ed. Side Effects of Drugs Annual. Amsterdam: Elsevier; 2017. (39): 269-282
- 8 Center For Drug Evaluation And Research. 209939Orig1s000; 209940Orig1s000. 2017
- 9 Lischka P, Hewlett G, Wunberg T. et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother 2010; 54 (03) 1290-1297
- 10 Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol 2011; 85 (20) 10884-10893
- 11 Imlay HN, Kaul DR. Letermovir and maribavir for the treatment and prevention of cytomegalovirus infection in solid organ and stem cell transplant recipients. Clin Infect Dis 2021; 73 (01) 156-160
- 12 Humphrey GR, Dalby SM, Andreani T. et al. Asymmetric synthesis of letermovir using a novel phase-transfer-catalyzed aza-michael reaction. Org Process Res Dev 2016; 20 (06) 1097-1103
- 13 Li R, Liu Z, Chen L, Pan J, Zhou W. Enantioselective phase-transfer catalyzed alkylation of 1-methyl-7-methoxy-2-tetralone: an effective route to dezocine. Beilstein J Org Chem 2018; 14: 1421-1427