
RSS-Feed abonnieren
DOI: 10.1055/s-0041-1740172
Inguinal Superficial CD34-Positive Fibroblastic Tumor. Case Report and Literature Review
Artikel in mehreren Sprachen: español | EnglishFunding The present research has not received any specific grant from public, commercial, or non-profit agencies.

Abstract
Superficial cluster of differentation 34 (CD34)-positive fibroblastic soft-tissue tumor is a rare, low-frequency tumor, characterized histologically by marked pleomorphism, low mitotic activity, and diffuse immunoreactivity for CD34. It may behave like a mesenchymal tumor of intermediate malignancy. There are only 52 cases published in the literature. We present the case of a 31-year-old patient with a long progressive and painful growth of a soft-tissue lesion in the left inguinal region. The mass was biopsied and, with the suspicion of a superficial CD34-positive fibroblast tumor, it was subsequently treated with an enlarged resection of the lesion and covering the skin defect with a local V-Y advancement flap, with a satisfactory evolution in the postoperative follow-up. The pathology report confirmed the diagnostic suspicion of a tumor with strong reactivity for CD34, with tumor protein p53 in 60% to 70%, antigen Ki67 in less than 15%, without loss of nuclear INI-1 protein, and with negativity for CD31, CD163, AE1AE3, CAM5.2, EMA, CD30, progestin receptors, S100 protein and desmin, with negative borders.
Level of evidence IV.
Keywords
CD34 - immunohistochemistry - mesenchymal tumors - superficial sarcoma - fibroblastic tumor - cytologyIntroduction
Superficial CD34-positive fibroblastic soft-tissue tumor is a rare condition, with 52 cases reported in the literature to date.[1] It is characterized by marked pleomorphism, low mitotic activity, CD34 immunoreactivity, and slow, progressive growth. It is believed to behave as a mesenchymal tumor of intermediate malignancy; however, due to the little information and scarce follow-up reports in the literature, the best treatment for this condition is yet to be determined. We present a case of a patient with a long-standing inguinal mass which was completely resected through surgery with adequate outcomes and no recurrences or complications during an 18-month clinical follow-up.
Case description
A 31-year-old female patient felt a mass in the left inguinal region for 10 years; it grew slowly and progressively, and started to cause pain in the proximal third of the left thigh in the previous year. Clinically, she presented a left inguinal, well-defined mass, close to the ischiopubic ridge, painful on palpation, with no skin or gait changes. Magnetic resonance imaging (MRI) scans revealed a well-defined, 3-cm x 4-cm lesion which was hypointense on T1-weighted and hyperintense on T2-weighted sequences, located on the medial aspect of the left thigh ([Fig. 1]).

Biopsy of the lesion was indicated for its characterization due to its size and the patient's pain complaints in recent months. The pathology report indicated the presence of a left inguinal mass with myxomatous features. The mass was positive for CD34, but negative for CD31, S100, SOX10, CK, desmin, signal transducer and activator of transcription 6 (STAT6) and estrogen receptors, among other markers. The characteristics of the lesion hindered its classification, and the mass was deemed a potential sarcoma.
The patient's case was presented to a medical board consisting of pathology, plastic surgery, and orthopedic oncology specialists, who decided for an en bloc resection of the lesion with widened margins and defect reconstruction with an advancement flap. The procedure was performed with no complications ([Fig. 2]).
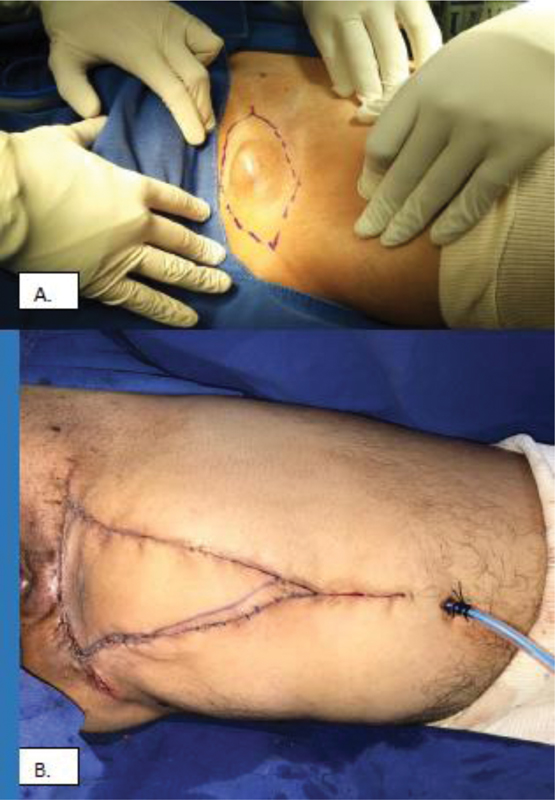
A gross pathology study revealed a superficial lesion, with well-defined edges, partially covered by skin ([Fig. 3]). A histopathology analysis revealed a lesion with sheets of pleomorphic cells mixed with clear xanthomatous cells, ([Fig. 4]) and no high mitotic count or necrosis; the lesion borders were negative for tumor. With these findings, several differential diagnoses were initially raised, including sarcomatoid carcinoma, melanoma, and atypical fibroxanthoma (FXA); however, there was no contact with the epidermis, however, there was no contact with the epidermis. Cytokeratins AE1AE3-CAM5.2, and proteins P.63, P.40, S100, SOX11, and MELAN A were negative, ruling out carcinoma and melanoma. Since the lesion occured on an area of skin with no sun exposure, FXA was ruled out; in addition, the mass was very large and deeply compromised. Diffuse CD34 reactivity ([Fig. 5]) broadened the diagnostic spectrum to include myxofibrosarcoma (which usually presents arcuate vessels and frequent mitoses in cases of high-grade lesions, as well as partial and not diffuse CD34 reactivity); vascular tumors, such as pleomorphic angiosarcoma (but there was no mitosis, the growth pattern was not infiltrative, and it was negative for Transmembrane E3 ubiquitin-protein ligase (FLY-1), cluster differentation 31 (CD31) and transcriptional regulator (ERG) were negative); and three related entities. The first was pleomorphic angiectatic tumor, but no ectatic vessels with peripheral fibrin deposits were found. The second one was myxoinflammatory fibroblastic sarcoma, but this is usually distally located and presents Reed-Sternberg-like cells and pseudolipoblasts; although it may exhibit CD34 reactivity, it is usually focal. The last differential diagnosis was a superficial CD34-positive fibroblastic tumor; no reactivity was found for desmin, AML or EMA, and there was no loss of INI-1. Fluorescent in situ hybridization for Transforming growth factor beta receptor type 3 (TGFBR3) or meningioma expressed antigen 5 (mgea5) was not available at our institution.



We concluded that this was a pleomorphic spindle cell tumor consistent with a superficial CD34-positive fibroblastic tumor.
During the 18-month clinical follow-up, the patient presented an adequate evolution, with proper wound healing and flap integration, in addition to no pain; there were no secondary complications or new lesions, and functionality was deemed adequate.
Discussion
Superficial CD34-positive fibroblastic tumor is rare and characterized by abundant pleomorphism, low mitotic activity, and CD34 immunoreactivity; it grows slowy and progressively. It usually occurs in the lower limbs, including the thigh, knee, buttocks, leg, and foot, and it is less frequent in other sites such as the arm, inguinal region, vulva, hip, shoulder, and neck. They are usually circumscribed lesions with subcutaneous presentation, with little or no deep-muscle involvement, as in our case.[1] [2] [3] [4] [5] [6] [7] [8] [9]
The first case was described by Carter et al.[3] in 2014, and, to date, 52 cases have been identified.[1] [2] [3] The literature presents only case reports, which makes it difficult to establish a treatment guide or clear evidence regarding surgical management and adjuvant modalities, such as chemotherapy or radiotherapy, for this type of tumor. However, the available information reveals good outcomes after surgical resection, with no postoperative local recurrences. The symptoms are highly variable, with disease onset up to 20 years before the surgical intervention.
As for pathophysiology, a behavior similar to that of a mesenchymal tumor of intermediate malignancy has been reported. The latest World Health Organization (WHO) classification of musculoskeletal tumors,[10] published in 2019, places this neoplasm within the group of fibroblastic/myofibroblastic tumors with intermediate, rarely metastatic behavior; rearrangements in domain zinc finger protein 10 (PFMR10) have been reported in some, and, so far, occasional cases with lymph node metastasis.
On histopathology, this tumor is characterized by pleomorphic, polygonal tumor cells oriented in sheets or fascicles, with abundant eosinophilic cytoplasm, hyperchromatic nuclei, and multiple nucleoli.[4] [6] Regarding immunohistochemistry, Lao et al.[6] report a strong, diffuse expression of CD34 at 100%, AE1/AE3 at 57%, and CAM5.2 at 50% with a low Ki67 index (1%) and negative findings for EMA, SMA, desmin, h-caldesmon, calponin, myogenin, myoblast determination protein 1 (MyoD1), S100 protein, transcription factor SOX-10, glial fibrillary acidic protein (GFAP), anti-melanoma monoclonal antibody HMB45, melan-A protein, CD31, ERG, STAT6 and anaplastic lymphoma kinase (ALK), findings that are consistent with those reported by Riddle et al.[4] and Li et al.[5] In 2014, in a series of 18 patients, Carter et al.[3] suggested that this entity must be considered a lesion of borderline malignancy.
Differential diagnoses include undifferentiated pleomorphic sarcoma, dermatofibrosarcoma protuberans, and myxofibrosarcoma.[3] [4] Correct differentiation of this type of tumor is critical for an adequate, timely treatment.
Conclusions
Superficial CD34-positive fibroblastic tumor is infrequent, with only 52 cases reported in the literature; it behaves as an intermediate-grade mesenchymal tumor, and its prognosis is excellent according to WHO[10] recommendations. Broader case series with long-term follow-ups are required to establish treatment protocols for this condition in terms of surgical management and potential adjuvant therapy.
Conflicto de Intereses
Los autores no tienen conflicto de intereses que declarar.
-
Referencías
- 1 Wada N, Ito T, Uchi H. et al. Superficial CD34-positive fibroblastic tumor: A new case from Japan. J Dermatol 2016; 43 (08) 934-936
- 2 Batur S, Ozcan K, Ozcan G, Tosun I, Comunoglu N. Superficial CD34 positive fibroblastic tumor: report of three cases and review of the literature. Int J Dermatol 2019; 58 (04) 416-422
- 3 Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27 (02) 294-302
- 4 Riddle NN, Gardner JM. The New Kids on the Block: Recently Characterized Soft Tissue Tumors. Surg Pathol Clin 2015; 8 (03) 467-491
- 5 Li W, Molnar SL, Mott M, White E, De Las Casas LE. Superficial CD34-positive fibroblastic tumor: Cytologic features, tissue correlation, ancillary studies, and differential diagnosis of a recently described soft tissue neoplasm. Diagn Cytopathol 2016; 44 (11) 926-930
- 6 Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70 (03) 394-401
- 7 Yamaga K, Fujita A, Osaki M. et al. Detailed analysis of a superficial CD34-positive fibroblastic tumor: A case report and review of the literature. Oncol Lett 2017; 14 (03) 3395-3400
- 8 Sood N, Khandelia BK. Superficial CD34-positive fibroblastic tumor: A new entity; case report and review of literature. Indian J Pathol Microbiol 2017; 60 (03) 377-380
- 9 Rekhi B, Banerjee D, Gala K, Gulia A. Superficial CD34-positive fibroblastic tumor in the forearm of a middle-aged patient: A newly described, rare soft-tissue tumor. Indian J Pathol Microbiol 2018; 61 (03) 421-424
- 10 Soft Tissue and Bone Tumours. WHO Classification of Tumours, 5th Edition, Volume 3. WHO Classification of Tumours Editorial Board. 2019
Address for correspondence
Publikationsverlauf
Eingereicht: 15. Mai 2020
Angenommen: 06. August 2021
Artikel online veröffentlicht:
22. Dezember 2021
© 2021. Sociedad Chilena de Ortopedia y Traumatologia. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commecial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Revinter Publicações Ltda.
Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
-
Referencías
- 1 Wada N, Ito T, Uchi H. et al. Superficial CD34-positive fibroblastic tumor: A new case from Japan. J Dermatol 2016; 43 (08) 934-936
- 2 Batur S, Ozcan K, Ozcan G, Tosun I, Comunoglu N. Superficial CD34 positive fibroblastic tumor: report of three cases and review of the literature. Int J Dermatol 2019; 58 (04) 416-422
- 3 Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27 (02) 294-302
- 4 Riddle NN, Gardner JM. The New Kids on the Block: Recently Characterized Soft Tissue Tumors. Surg Pathol Clin 2015; 8 (03) 467-491
- 5 Li W, Molnar SL, Mott M, White E, De Las Casas LE. Superficial CD34-positive fibroblastic tumor: Cytologic features, tissue correlation, ancillary studies, and differential diagnosis of a recently described soft tissue neoplasm. Diagn Cytopathol 2016; 44 (11) 926-930
- 6 Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70 (03) 394-401
- 7 Yamaga K, Fujita A, Osaki M. et al. Detailed analysis of a superficial CD34-positive fibroblastic tumor: A case report and review of the literature. Oncol Lett 2017; 14 (03) 3395-3400
- 8 Sood N, Khandelia BK. Superficial CD34-positive fibroblastic tumor: A new entity; case report and review of literature. Indian J Pathol Microbiol 2017; 60 (03) 377-380
- 9 Rekhi B, Banerjee D, Gala K, Gulia A. Superficial CD34-positive fibroblastic tumor in the forearm of a middle-aged patient: A newly described, rare soft-tissue tumor. Indian J Pathol Microbiol 2018; 61 (03) 421-424
- 10 Soft Tissue and Bone Tumours. WHO Classification of Tumours, 5th Edition, Volume 3. WHO Classification of Tumours Editorial Board. 2019




