Subscribe to RSS
DOI: 10.1055/s-0041-1740172
Tumor fibroblástico inguinal superficial positivo para CD34. Reporte de un caso y revisión de la literatura
Article in several languages: español | EnglishAuthors
Financiamiento La presente investigación no ha recibido ninguna beca específica de agencias de los sectores público, comercial, o sin ánimo de lucro.

Resumen
El tumor fibroblástico superficial de tejidos blandos positivo para antígeno CD34 (CD34) es un tumor raro, de baja frecuencia, que se caracteriza histológicamente por un marcado pleomorfismo, baja actividad mitótica, e inmunoreactividad difusa para CD34. Puede tener un comportamiento similar al de un tumor mesenquimal de malignidad intermedia. Existen sólo 52 casos publicados en la literatura. Se presenta el caso de una paciente de 31 años con una masa en tejidos blandos en región inguinal izquierda, de crecimiento progresivo, de varios meses de evolución dolorosa. La masa fue biopsiada y, con la sospecha de un tumor fibroblástico superficial positivo para CD34, fue posteriormente tratada con una resección ampliada de la lesión y con cobertura del defecto en la piel con un colgajo local de avance de V-Y, con una evolución satisfactoria en su seguimiento postquirúrgico. El reporte de patología confirmó la sospecha diagnóstica de un tumor con reactividad fuerte para CD34, con proteína P53 en 60% a 70%, antígeno Ki67 menor al 15%, sin pérdida de proteína nuclear INI-1, y negatividad para CD31, CD163, AE1AE3, CAM5.2, EMA, CD30, receptores de progestágenos, proteína S100, y desmina, con bordes negativos.
Nivel de evidencia IV.
Palabras Clave
CD34 - inmunohistoquímica - tumores mesenquimales - sarcoma superficial - tumor fibroblástico - citologíaIntroducción
El tumor fibroblástico superficial de tejidos blandos positivo para CD34 es un tumor raro, de baja frecuencia, y se han reportado en la literatura 52 casos hasta la fecha.[1] Se caracteriza por marcado pleomorfismo, con baja actividad mitótica e inmunoreactividad a CD34, con un crecimiento lento y progresivo. Se considera que puede tener un comportamiento similar al de un tumor mesenquimal de malignidad intermedia; sin embargo, debido a la poca información y seguimientos existentes en la literatura, no existe claridad en cuanto al mejor manejo disponible para esta patología. Se presenta un caso de una paciente con una masa inguinal de larga data, resecada completamente mediante cirugía con adecuado resultado, sin recidiva ni complicaciones durante su seguimiento clínico a 18 meses.
Descripción del Caso
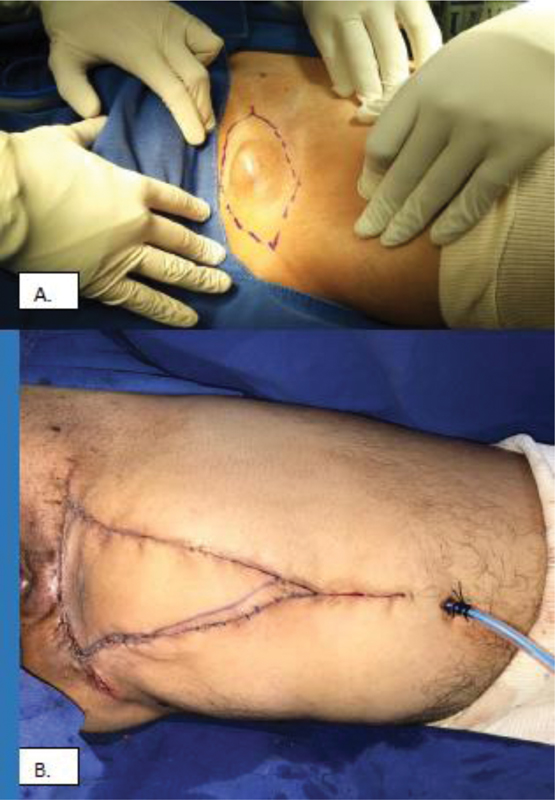
Paciente femenina de 31 años, con sensación de masa en la región inguinal izquierda, de 10 años de evolución, de crecimiento lento, progresivo, con dolor en el tercio proximal del muslo izquierdo en el último año. Clínicamente, presenta una masa en la región inguinal izquierda, cercana al reborde isquiopubiano, dolorosa a la palpación, bien definida, sin cambios cutáneos, sin alteración de la marcha. Se realizó una resonancia nuclear magnética (RNM), en la que se identificó una lesión hipointensa en T1 e hiperintensa en T2, bien definida, de 3 cm x 4 cm, ubicada en la cara medial del muslo izquierdo ([Fig. 1]).

Se indicó la toma de una biopsia de la lesión, dado su tamaño y el dolor referido en los últimos meses, para su caracterización. El reporte de patología indicó la presencia de una masa en la región inguinal izquierda, de características mixomatosas. Se encontró positividad para CD34 y negatividad para CD31, S100, SOX10, CK, desmina, transductor de señal y activador de transcripción 6 (STAT6), y receptor de estrógenos, entre otros. Dadas las características de la lesión, hubo dificultad para su clasificación, y se rotuló como un posible sarcoma.
Se presentó el caso de la paciente en una junta médica en conjunto con patología, cirugía plástica, y ortopedia oncológica, y se decidió realizar una resección en bloque de la lesión con márgenes ampliados y reconstrucción del defecto con un colgajo de avance, procedimiento realizado sin complicaciones ([Fig. 2]).

El estudio de patología reportó macroscópicamente una lesión superficial de bordes bien definidos recubierta parcialmente por piel ([Fig. 3]). En histopatología, se encontró una lesión con sábanas de células pleomórficas mezcladas con células claras xantomatosas ([Fig. 4]), sin conteo mitótico alto ni necrosis, y con bordes negativos para tumor. Con estos hallazgos, se plantearon varios diagnósticos diferenciales, que incluyeron: carcinoma sarcomatoide, melanoma, y fibroxantoma atípico (FXA), inicialmente; sin embargo, no hubo contacto con la epidermis, y las citoqueratinas AE1AE3 - CAM5.2 y las proteínas P.63, P.40, S100, SOX11 y MELAN A fueron negativas, lo que nos hizo descartar el diagnóstico de carcinoma y melanoma. En cuanto al FXA, esta lesión no está en piel expuesta al sol, es muy grande, y comprometió profundamente; por lo tanto, se descartó. Se encontró reactividad difusa para CD34 ([Fig. 5]), lo que amplió el espectro de diagnóstico, incluyendo: mixofibrosarcoma (pero usualmente hay vasos arqueados y mitosis frecuentes cuando son de alto grado, y reactividad parcial y no difusa para CD34); un tumor vascular, como angiosarcoma pleomórfico, pero no hay mitosis, el patrón de crecimiento no fue infiltrativo, y hubo negatividad para proteína FLY.1, antígeno CD31, regulador transcripcional ERG; y tres entidades relacionadas, que son el tumor angiectásico pleomórfico (pero no se encontraron vasos ectásicos con depósitos periféricos de fibrina), el sarcoma fibroblástico mixoinflamatorio (pero este suele ser distal, con células similares a las de Reed-Sternberg y pseudolipoblastos; adicionalmente está descrita reactividad para CD34, pero suele ser focal), y, por último, el que se favoreció: el tumor fibroblástico superficial positivo para CD34. No se encontró reactividad para desmina, AML y EMA, y no hubo pérdida de INI-1. En nuestra institución, no hay disponibilidad de FISH para TGFBR3 o factor transformador del receptor del factor de crecimiento beta tipo antígeno MGEA5.



Se concluyó que se trataba de un tumor fusocelular y pleomórfico compatible con tumor fibroblástico superficial positivo para CD34.
Durante el seguimiento clínico a 18 meses, la paciente presentó una evolución adecuada sin dolor, con adecuada cicatrización de la herida e integración del colgajo, sin complicaciones secundarias ni presencia de nuevas lesiones, y con adecuada funcionalidad.
Discusión
El tumor fibroblástico superficial positivo para CD34 es raro, de baja ocurrencia, caracterizado por abundante pleomorfismo, baja actividad mitótica e inmunoreactividad a CD34, el cual tiene un crecimiento lento y progresivo. Generalmente se presenta en miembros inferiores: muslo, rodilla, glúteos, pierna y pie, y, en menor frecuencia, en otras localizaciones, incluyendo brazos, región inguinal, vulva, cadera, hombro y cuello. Suelen ser lesiones circunscritas, y se presentan de manera subcutánea, con poca o nula afección de la musculatura profunda, como lo descrito en nuestro caso.[1] [2] [3] [4] [5] [6] [7] [8] [9]
EL primer caso fue descrito por Carter et al.[3] en 2014, y hasta la fecha se han identificado 52 casos.[1] [2] [3] [4] En la literatura, sólo existen reportes de caso, lo cual dificulta que se cuente con una guía de manejo o evidencia clara en cuanto al manejo quirúrgico y adyuvante con quimio o radioterapia para este tipo de tumor; sin embargo, la información disponible reporta un buen resultado con la resección quirúrgica. En las series de casos y reportes, se ha encontrado que ninguno presentó recurrencia local posterior a la resección quirúrgica. La presentación de los síntomas de los pacientes es muy variable, y hay reportes con inicio hasta 20 años antes de la intervención quirúrgica.
En cuanto a su fisiopatología, se ha reportado un comportamiento similar al de un tumor mesenquimal de malignidad intermedia. En la última clasificación de tumores musculoesqueléticos de la Organización Mundial de la Salud[10] (OMS), del 2019, esta neoplasia está dentro del grupo de tumores fibroblásticos/miofibroblásticos de comportamiento intermedio, rara vez metastatizante; rearreglos en proteína PRDM10 han sido reportados en algunos casos y, hasta el momento, ocasionales casos con metastasis ganglionar.
En histopatología, se caracteriza por células tumorales pleomórficas y poligonales, las cuales pueden orientarse en forma de sábanas o fascículos, con abundante citoplasma eosinofílico y núcleos con hipercromasia y múltiples nucleolos.[4] [6] En cuanto a la inmunohistoquímica, en su serie, Lao et al.[6] reportan una expresión fuerte y difusa de CD34 del 100%, AE1/AE3 en 57%, y CAM5.2 en el 50%, con un índice de Ki67 bajo (1%) y negatividad para EMA, SMA, desmina, h-caldesmon, calponina, miogenina, Proteína de determinación de mioblastos 1 (MyoD1), S100, factor de transcripción SOX-10, proteína ácida fibrilar glial (GFAP), anticuerpo monoclonal anti-melanoma HMB45, proteína melan-A, CD31, proteína ERG, STAT6, y cinasa de linfoma anaplásico ALK, hallazgos que concuerdan con los reportados por Riddle et al.[4] y Li et al.[5] En 2014, Carter et al.,[3] en una serie de 18 pacientes, sugirieron que esta entidad se considere como una lesión de malignidad límite.
Entre los diagnósticos diferenciales, se consideran: sarcoma pleomórfico indiferenciado, dermatofibrosarcoma protuberans, y mixofibrosarcoma.[3] [4] Es fundamental la diferenciación correcta de este tipo de tumor a fin de poder realizar un tratamiento adecuado y oportuno.
Conclusiones
El tumor fibroblástico superficial positivo para CD34 es infrecuente, con apenas 52 casos reportados en la literatura. Tiene un comportamiento similar al de un tumor mesenquimal de grado intermedio, y se ha reportado un pronóstico excelente con base en las recomendaciones de la OMS.[10] Se requieren series de casos más amplias y seguimientos a largo plazo para poder establecer para esta patología protocolos de manejo quirúrgico y la posibilidad de manejo adyuvante.
Conflicto de Intereses
Los autores no tienen conflicto de intereses que declarar.
-
Referencías
- 1 Wada N, Ito T, Uchi H. et al. Superficial CD34-positive fibroblastic tumor: A new case from Japan. J Dermatol 2016; 43 (08) 934-936
- 2 Batur S, Ozcan K, Ozcan G, Tosun I, Comunoglu N. Superficial CD34 positive fibroblastic tumor: report of three cases and review of the literature. Int J Dermatol 2019; 58 (04) 416-422
- 3 Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27 (02) 294-302
- 4 Riddle NN, Gardner JM. The New Kids on the Block: Recently Characterized Soft Tissue Tumors. Surg Pathol Clin 2015; 8 (03) 467-491
- 5 Li W, Molnar SL, Mott M, White E, De Las Casas LE. Superficial CD34-positive fibroblastic tumor: Cytologic features, tissue correlation, ancillary studies, and differential diagnosis of a recently described soft tissue neoplasm. Diagn Cytopathol 2016; 44 (11) 926-930
- 6 Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70 (03) 394-401
- 7 Yamaga K, Fujita A, Osaki M. et al. Detailed analysis of a superficial CD34-positive fibroblastic tumor: A case report and review of the literature. Oncol Lett 2017; 14 (03) 3395-3400
- 8 Sood N, Khandelia BK. Superficial CD34-positive fibroblastic tumor: A new entity; case report and review of literature. Indian J Pathol Microbiol 2017; 60 (03) 377-380
- 9 Rekhi B, Banerjee D, Gala K, Gulia A. Superficial CD34-positive fibroblastic tumor in the forearm of a middle-aged patient: A newly described, rare soft-tissue tumor. Indian J Pathol Microbiol 2018; 61 (03) 421-424
- 10 Soft Tissue and Bone Tumours. WHO Classification of Tumours, 5th Edition, Volume 3. WHO Classification of Tumours Editorial Board. 2019
Address for correspondence
Publication History
Received: 15 May 2020
Accepted: 06 August 2021
Article published online:
22 December 2021
© 2021. Sociedad Chilena de Ortopedia y Traumatologia. This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commecial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Thieme Revinter Publicações Ltda.
Rua do Matoso 170, Rio de Janeiro, RJ, CEP 20270-135, Brazil
-
Referencías
- 1 Wada N, Ito T, Uchi H. et al. Superficial CD34-positive fibroblastic tumor: A new case from Japan. J Dermatol 2016; 43 (08) 934-936
- 2 Batur S, Ozcan K, Ozcan G, Tosun I, Comunoglu N. Superficial CD34 positive fibroblastic tumor: report of three cases and review of the literature. Int J Dermatol 2019; 58 (04) 416-422
- 3 Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol 2014; 27 (02) 294-302
- 4 Riddle NN, Gardner JM. The New Kids on the Block: Recently Characterized Soft Tissue Tumors. Surg Pathol Clin 2015; 8 (03) 467-491
- 5 Li W, Molnar SL, Mott M, White E, De Las Casas LE. Superficial CD34-positive fibroblastic tumor: Cytologic features, tissue correlation, ancillary studies, and differential diagnosis of a recently described soft tissue neoplasm. Diagn Cytopathol 2016; 44 (11) 926-930
- 6 Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology 2017; 70 (03) 394-401
- 7 Yamaga K, Fujita A, Osaki M. et al. Detailed analysis of a superficial CD34-positive fibroblastic tumor: A case report and review of the literature. Oncol Lett 2017; 14 (03) 3395-3400
- 8 Sood N, Khandelia BK. Superficial CD34-positive fibroblastic tumor: A new entity; case report and review of literature. Indian J Pathol Microbiol 2017; 60 (03) 377-380
- 9 Rekhi B, Banerjee D, Gala K, Gulia A. Superficial CD34-positive fibroblastic tumor in the forearm of a middle-aged patient: A newly described, rare soft-tissue tumor. Indian J Pathol Microbiol 2018; 61 (03) 421-424
- 10 Soft Tissue and Bone Tumours. WHO Classification of Tumours, 5th Edition, Volume 3. WHO Classification of Tumours Editorial Board. 2019