

Subscribe to RSS
DOI: 10.1055/s-0041-1727235
Supramolecular Systems Containing B–N Frustrated Lewis Pairs of Tris(pentafluorophenyl)borane and Triphenylamine Derivatives
Authors

Abstract
The introduction of a chemical additive to supramolecular polymers holds high potential in the development of new structures and functions. In this regard, various donor- and acceptor-based molecules have been applied in the design of these noncovalent polymers. However, the incorporation of boron–nitrogen frustrated Lewis pairs in such architectures is still rare despite their many intriguing properties in catalysis and materials science. The limited choices of suitable boron derivatives represent one of the main limitations for the advancement in this direction. Here, we examine the use of the commercially available tris(pentafluorophenyl)borane with various triphenylamine derivatives to create supramolecular B–N charge transfer systems. Our results highlight the importance of a proper balance between the donor/acceptor strength and the driving force for supramolecular polymerization to achieve stable, long-range ordered B–N systems. Detailed analyses using electron paramagnetic resonance and optical spectroscopy suggest that tris(pentafluorophenyl)borane displays complex behavior with the amide-based triphenylamine supramolecular polymers and may interact in dimers or larger chiral aggregates, depending on the specific structure of the triphenylamines.
Key words
supramolecular assembly - tris(pentafluorophenyl)borane - triphenylamine - charge transferIntroduction
Incorporating multiple components in an assembly process is a promising approach to achieve new structures, properties and hence functions.[1] [2] This strategy has been widely applied in the field of supramolecular polymers in the last decades with the aim of exploring and understanding unprecedented behavior and properties of supramolecular polymers.[3] [4] The interplay between components possessing fundamentally different interaction modes can lead to phenomena that are otherwise challenging to achieve, such as dilution-induced self-assembly,[5] thermally bisignate supramolecular polymerization[6] and sequence-controlled supramolecular copolymerization.[7] The nature of the hetero-interactions between different components can greatly determine the supramolecular microstructure and the corresponding properties. Among many supramolecular interactions, the donor–acceptor interaction is one of the most promising tools to design alternating copolymers, since by nature the homo-interactions are repulsive and the hetero-interactions are attractive.[8] This design principle has been used successfully in donor–acceptor oligomers and polymers by Peter Bäuerle and his group.[9]
Boron–nitrogen interactions have become the basis in designing a wide range of luminescent materials.[10] [11] Due to their donor–acceptor nature, the nitrogen and boron moieties can form a dative B–N bond, either in an intra- or intermolecular fashion.[12] [13] In contrast, if the steric congestion around the B and N centers hinders dative bond formation, a non-covalent transient dimer, known as frustrated Lewis pair (FLP), can form. The latter has been explored in many applications ranging from catalysis,[14] optoelectronics[15] [16] to stimuli-responsive materials.[17] [18] However, examples that incorporate B–N FLPs in supramolecular polymer systems are still rare.[19]
We recently reported supramolecular block-like copolymers based on discotic chiral triarylamine and triarylborane building blocks.[19] B–N contacts in the copolymers generated B–N charge transfer, leading to a circularly polarized charge transfer luminescence. Mechanistic studies revealed that, despite the high similarity of the structures, only a small fraction of B–N contacts was formed in solution. This was most likely caused by strong competition between the noncovalent forces to form a polymer and the B–N interactions.[19] Thus, designing a stronger B–N pair will be highly beneficial to increase the degree of B–N mixing, hereby enhancing the effect of B–N interaction. Taking inspiration from the classic triphenylborane–triphenylamine FLP, we wonder whether a more simple B–N dimer can be extended into a one-dimensional (1D) array, with the aim to achieve long-range ordering for optoelectronic applications.[20]
Here, we report the synthesis and characterization of B–N based supramolecular systems. Various derivatives of triphenylamine (TPA) were synthesized ([Scheme 1]) and characterized for their interactions with tris(pentafluorophenyl)borane (TFPB, [Scheme 1]), which is commercially available and bypasses the limitation in synthetic accessibility of boron compounds. TFPB was chosen due to its higher Lewis acidity and better resistance to air and moisture in comparison to other boron derivatives such as triphenylborane and trimesitylborane. By utilizing TFPB, we aim to generalize the use of B–N FLPs in supramolecular systems.

Results and Discussion
Design and Synthesis of Triphenylamine Derivatives
Four different donor molecules based on the TPA core were designed with the capability to form supramolecular polymers, but which differ in electron density and nature of the non-covalent interactions. First, we extended the conjugation system and increased the electron density of the nitrogen central atom by attaching electron-donating groups in the side chains (TPA-alkyne, [Scheme 1]). TPA-alkyne contains a large π-system, which potentially contributes to the aggregation tendency. We speculate that, in the presence of TFPB, an arene–perfluoroarene interaction could also aid in promoting the formation of B–N contacts in this B–N system.[21] [22] The synthesis of TPA-alkyne was carried out in one step via a Sonogashira cross-coupling reaction between tris(4-iodophenyl)amine and (3,4,5-tridodecyloxyphenyl)acetylene. The product was obtained in 76% yield as a light-yellow solid.
The non-planar geometry of TPA-alkyne around the nitrogen center, in combination with N–arene and dodecyl C–C bond rotational freedom, could reduce the effect of the intermolecular π-stacking.[23] Such counter-effects might limit the assembly ability of TPA-alkyne. Thus, to deliberately promote the assembly of TPA monomers, we incorporated amide units as the linkers between the TPA core and the side chains in triphenylamine triamide (TPA-amide, [Scheme 1]). This TPA derivative can form 1D supramolecular polymers in nonpolar solvents mediated by the intermolecular H-bonding network.[24] TPA-amide was obtained as a white solid in 36% yield from the amidation reaction between 4,4′,4′′-nitrilotribenzoic acid and (S)-3,7-dimethyloctylamine.
An intercalation of TFPB in TPA-amide polymers could increase the intermolecular distance between TPA-amide monomers, thus potentially interfering with their H-bonding network. To reduce this effect, we designed triphenylamine nona-amide (TPA-amide9, [Scheme 1]) which contains the glutamic acid-based side chains that allow the formation of a multimodal H-bonding network in both inter- and intramolecular manners.[25] We hypothesize that this branched-amide strategy could maintain a H-bonding network while at the same time allow TFPB to intercalate. TPA-amide9 was synthesized in three steps, starting from an amidation reaction between t-Boc-protected l-glutamic acid and two dodecylamines, followed by t-Boc deprotection under acidic conditions.[26] [27] The target molecule was synthesized by coupling this (S)-2-amino-N 1,N 5-didodecylpentanediamide with 4,4′,4′′-nitrilotribenzoic acid to obtain a white solid in 79% yield. Comparing to TPA-alkyne, these two amide-based TPA derivatives are less electron rich due to the presence of electron-withdrawing groups.
Lastly, we replaced the amide linkers with the esters in the design of triphenylamine triester TPA-ester ([Scheme 1]) to study the role of the hydrogen bonds. The synthesis of TPA-ester was carried out in one step via the esterification between 4,4′,4′′-nitrilotribenzoic acid and (S)-3,7-dimethyloctanol, yielding a colorless, viscous liquid in 32% yield. All TPA derivatives were fully characterized by 1H NMR, 13C NMR and mass spectrometry.
Photoinduced B–N Electron Transfer
Before studying the possibility of supramolecular B–N assembly, we first investigated the presence and nature of the interaction between TFPB and TPA derivatives by electron paramagnetic resonance (EPR) spectroscopy.[28] TPA-alkyne/TFPB and TPA-amide/TFPB mixtures (c TPA , c TFPB = 500 µM) were prepared in a degassed 9:1 v/v decalin (Dec)/1,2-dichloroethane (DCE) solvent mixture to promote the interaction between the components. X-band EPR spectra recorded at 20 K for TPA-alkyne/TFPB and 9 K for TPA-amide/TFPB display two distinct signals with g–values of 2.0 and 4.2 during light irradiation (λex = 320–390 nm), whereas no resonances were observed prior to photoexcitation ([Figure 1] for TPA-alkyne/TFPB and [Figure S1] for TPA-amide/TFPB). The appearing resonances during excitation indicate the formation of radical species resulting from single-electron transfer from TPA to TFPB in these B–N mixtures. Whereas the signal observed at g = 2.0 is characteristic for a spin doublet system (S = ½; one unpaired electron) located at an organic molecule (i.e., likely B or N centered), the signal observed during irradiation at g = 4.2 is indicative of a spin triplet (S = 1) system (i.e., two coupled unpaired α-spin electrons in close proximity), most likely the result of the N-to-B single-electron transfer.[29] In absence of an excitation source, the g = 4.2 signal diminishes, while the intensity of the signal at g = 2.0 is reduced significantly. As the spin triplet species (g = 4.2) is only populated under constant excitation, this indicates reversible single-electron transfer between the B–N (ground state) and B•−–N•+ (excited state) FLP at low temperatures.[28] However, the more persistent resonance at g = 2.0 indicates that to some degree a small number of isolated radicals (anions/cations) are formed during the excitation process at low temperatures.

Having confirmed the B–N electron transfer process at low temperatures, we further investigated the interactions between TPA and TFPB by optical spectroscopy. No interaction between TPA-alkyne and TFPB is observed in the Dec/DCE solvent mixture at room temperature ([Figure S2a, b]). We serendipitously observed that mixing TPA-alkyne and TFPB in chlorinated solvents causes a rapid change of the mixture's color from slightly yellow to dark green, in which the intensity of the green color increases over several minutes ([Figure S3]). Further investigations into this unexpected color change show that the photoluminescence spectra of the aged 1:1 mixture (c TPA-alkyne , c TFPB = 2 mM, chloroform) has an emission peak at λem = 413 nm and a new band at λem ≈ 502 nm, whereas pure TPA-alkyne displays only one emission peak at λem = 413 nm ([Figures S2c, d]). The emission band at 502 nm can arise from a charge transfer excited state of a B–N complex.[15] [19] This emission wavelength is an indicator of the energy of the charge transfer interaction and can be correlated to the energetic difference between the donor's HOMO and the acceptor's LUMO. The broadening and disappearance of peaks located in the aromatic regions of 1H and 13C NMR spectra of this B–N mixture in deuterated chloroform further support the complexation between TPA-alkyne and TFPB ([Figure S4]). However, the sample loses its emission after prolonged equilibration time at room temperature ([Figure S2d]). The formation of green species and fluorescence quenching are typically related to the formation of triphenylamine radical cations.[30] [31] It is known that TFPB can form a radical anion that decomposes rapidly at room temperature.[32] [33] This situation may occur with TPA-alkyne/TFPB, leaving TPA-alkyne in its radical cationic state. Although a strong donor–acceptor strength is beneficial to achieve a high degree of mixing, the drawback is the irreversibility of the charge transfer process at room temperature which leads to an instability of the complex.
We proceeded with the TPA-ester which is less electron-rich than TPA-alkyne. Adding 1 equiv of TFPB to TPA-ester (c TPA-ester = 25 µM, 9:1 v/v Dec/DCE) results in a negligible difference in both absorption and emission spectra ([Figures S5b, c]), compared to pure TPA-ester. When TFPB is added in excess, we observed clear changes in the absorption and emission spectra ([Figures S5b, c]). The emission of TPA-ester at λem = 380 nm is quenched, and a weak emission band appears at λem = 520 nm, which can be ascribed to the formation of TPA-ester/TFPB charge transfer excited states. However, circular dichroism (CD) measurements indicate no sign of chiral long-range assemblies at room temperature ([Figure S5a]). Thus, we conclude that the B–N interaction between TPA-ester and TFPB is not strong enough to promote the formation of extended B–N assemblies in this B–N system. In addition, the homo-interactions between TPA-ester monomers are not sufficient to promote the supramolecular homopolymerization of TPA-ester as probed by CD measurement ([Figure S5a]).
Incorporation of H-Bonding as an Additional Driving Force for B–N Aggregates
The assembly of TPA-amide in nonpolar solvents was previously reported.[24] Thus, we first studied the assembly of the new TPA derivative TPA-amide9. CD measurements of TPA-amide9 in 9:1 v/v Dec/DCE (c TPA-amide9 = 50 µM) indicate the formation of helical structures with preferred handedness ([Figure S6a]), which is in agreement with the fibrous structures revealed by atomic force microscopy ([Figure S7]). TPA-amide9 also aggregates in chloroform ([Figure S8]), while no assembly is observed for TPA-amide under the same conditions. These results can be attributed to the presence of multiple amide units in TPA-amide9 that strengthen the H-bonding network in both inter- and intramolecular manners.[25] Indeed, TPA-amide9 aggregates display high thermal stability which do not fully dissociate at 80 °C in the Dec/DCE solvent mixture ([Figure S6a], red line). As a result, CD spectra of TPA-amide9 at 20 °C after cooling from 80 °C can vary in shape and intensity (data not shown). For photophysical properties, TPA-amide9 polymers in the Dec/DCE solvent mixture show the maximum emission at λem = 395 nm ([Figure 2a], red line), which is slightly red-shifted in comparison to TPA-amide ([Figure 2b], red line). Similarly, the assembly of TPA-amide9 can also be observed in its photoluminescence spectra at high temperatures ([Figure S6c], red line). Hence it is important to note that the assembly product of TPA-amide9 is, unlike TPA-amide, not completely in thermodynamic equilibrium under our experimental conditions.

The interactions between TPA-amide9/TPA-amide and TFPB were then evaluated by photoluminescence experiments. Both TPA derivatives at a concentration of 50 μM were mixed with 0–1 equiv of TFPB in the degassed Dec/DCE solvent mixture. All mixtures were heated to 80 °C and cooled to 20 °C with the increments of 10 °C followed by an equilibration for 10 minutes. The photoluminescence studies show that the 1:1 TPA-amide/TFPB mixture displays a significant quenching of the TPA-amide emission at λem = 390 nm and the appearance of a new broad emission band at λem = 442 nm ([Figure 2b], blue line), which is in line with the photoinduced electron transfer as probed by X-band EPR ([Figure S1]). Similarly, a new emission band at λem = 485 nm is observed in the TPA-amide9/TFPB mixture ([Figure 2a], blue line). It is of note that adding 0.5 equiv of TFPB already generates an intense charge transfer luminescence at 20 °C ([Figure 2a, b], yellow lines). These B–N charge transfer emissions are blue-shifted when compared to the TPA-ester/TFPB system ([Figure S5c]).
Comparing the two TPA derivatives and their interactions with TFPB shows that the emission band of TPA-amide9/TFPB systems is more red-shifted, suggesting two possibilities: 1) TPA-amide9 is a stronger donor than TPA-amide or 2) the intermolecular distance between B and N is shorter in the case of TPA-amide9 than with TPA-amide.[34] [35] [36] Moreover, the TPA-amide9/TFPB system displays a lower charge transfer/homopolymer emission intensity ratio than the TPA-amide/TFPB mixtures. This is likely caused by a lower number of B–N contacts in TPA-amide9/TFPB systems due to high thermal stability of TPA-amide9 polymers that could hinder the interactions with TFPB. In addition, the emission of B–N mixtures in solution is also blue-shifted in comparison to their bulk samples (λem = 458 nm for TPA-amide/TFPB and λem = 495 nm for TPA-amide9/TFPB, [Figure S9]), which can be attributed to a longer intermolecular distance between the B and N molecules in solution.[34] [36] All together, these results point to the different nature of the interactions between TPA-amide/TPA-amide9 and TFPB.
To gain more insight into the charge transfer emission, we recorded the emission spectra of the amide-based TPA derivatives and TFPB mixtures at different temperatures. At 80 °C, the 1:1 TPA-amide/TFPB mixture shows main emission peaks at λem = 382 and λem ∼ 450 nm, in which the latter is assigned to B–N charge transfer ([Figure 2c], red line). In contrast, the mixture containing 0.5 equiv of TFPB shows only one peak at λem = 377 nm ([Figure S10], red line), indicating that B–N interactions are not prevalent at this high temperature when only 50% TFPB is present. These results suggest different interaction modes between TPA-amide and TFPB when using different TFPB concentrations. For TPA-amide9/TFPB mixtures, the intense charge transfer emission can be already observed at 0.5 equiv of TFPB ([Figure S11a], red line). This situation is most likely mediated by high thermal stability of TPA-amide9 aggregates that efficiently trap TFPB and enhance B–N interactions. Decreasing the temperature quenches and red-shifts the emission of both TPA derivatives, which could be caused by polymerization and/or charge transfer to TFPB ([Figures 2c], [S10] and [S11], blue lines).[19] [24] This trend is accompanied by the growth of the charge transfer emission, where the red-shift is an indication of a shorter distance between B and N components in the mixed systems.[34] [36]
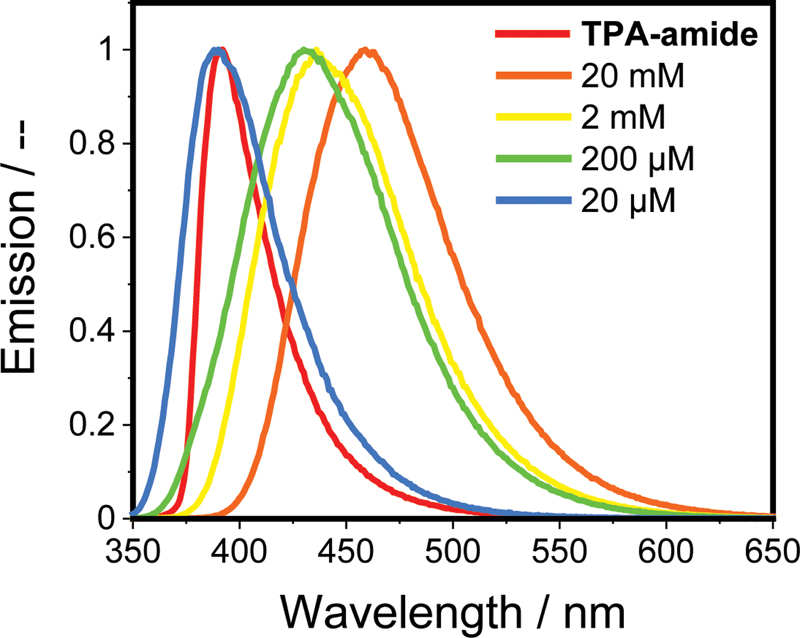
Both amide-based TPA derivatives form stable gels in decalin (20 mM for TPA-amide and 2 mM for TPA-amide9) and emit purple/blue light when excited with light of λex = 365 nm. Adding TFPB leads to a fluent, light-blue emitting solution ([Figure S12] for TPA-amide gel). [Figure 3] shows the normalized emission spectra of TPA-amide/TFPB mixtures of different concentrations. The wavelength of the onset of emission undergoes a blue-shift upon dilution. Interestingly, the 20 mM sample has a lower energy maximum than the 2 mM and 200 μM solutions. This wavelength shift could be due to an increase in distance between B and N upon dilution, which reduces supramolecular aggregation and causes the charge transfer complex to emit light of a higher energy.[34] [36] Diluting the sample further to 20 μM results in a similar maximum emission compared to pure TPA-amide, indicating the recovery of TPA-amide homopolymers and the absence of B–N interactions at this concentration regime. In contrast, the emission of the TPA-amide9/TFPB mixture is stable over a wide range of concentrations, further confirming the higher stability of TPA-amide9/TFPB aggregates ([Figure S13a]). In both cases, the reemerging emission of the monomers at low B–N mixture's concentration highlights that the formation of TPA/TFPB charge transfer complexes is reversible. No irreversible covalent-bond formation causes the new emission band, which was also confirmed by mass spectrometry ([Figures S13b] and [S14]).
Finally, we performed circularly polarized luminescence (CPL) measurements to probe whether the charge-transfer excited state is a loose dimer or rather a geometrically restricted, chiral aggregate. [Figure 4] shows that the TPA-amide9/TFPB charge transfer luminescence is circularly polarized with a dissymmetry factor (g LUM) of ∼6 × 10−4 at λex = ∼500 nm, which is in a typical regime for organic molecules (see [Figure S15] for additional CPL of TPA-amide9).[37] In contrast, the emission of TPA-amide/TFPB is not circularly polarized ([Figure S16]). These results suggest that TFPB interacts differently with the two amide-based TPA derivatives. It is very likely that TPA-amide9 and TFBP in the B–N pair stack on top of each other with a twisted orientation to allow the electron movement from the donor to the acceptor in a helical path. Thus, the B–N contact in the TPA-amide9/TFPB system are not merely loose dimers (which should be the case for TPA-amide/TFPB) but must have a helical organization that will generate the circular polarized charge transfer luminescence. Aggregates of TPA-amide9 could retain the helical order in the H-bonding network while allowing TFPB to intercalate when present in various concentrations as indicated by the active CD peaks ([Figure S17a]). In contrast, the helical ordering of TPA-amide aggregates is significantly affected in the presence of TFPB (see below).

Analysis of the Possible Interaction Modes between TPA-Amide and TFPB
A combination of spectroscopic techniques and theoretical simulation was used to study how TFPB affects the supramolecular polymerization of TPA-amide such as a chain capper, intercalator or sequestrator ([Figure 5]).[38] Due to its high thermal stability, we note that TPA-amide9 was not further investigated with these techniques.

As TPA-amide can form chiral polymeric stacks in solution, CD spectroscopy can be used to probe any changes after introducing TFPB to TPA-amide solutions ([Figure 6a]). CD spectra for all mixtures at 20 °C are similar in shape but the intensity of the main CD peak is lower in the presence of TFPB. This decrease in optical activity indicates a lower structural order (i.e., helicity in our case) and can be caused by the depolymerization of TPA-amide homopolymer stacks, which could be confirmed by the red-shifted main absorption peak in the presence of TFPB ([Figure S18a]). As pure TPA-amide polymers display a blue-shifted absorption peak upon polymerization, the observed redshift indicates the lower degree of polymerization of TPA-amide in these B–N mixtures.[24]

We then monitored the change of CD and absorption signals as a function of temperature ([Figure 6b] for CD and [Figure S18b] for UV-Vis). CD cooling curves show that TPA-amide homopolymers (c TPA-amide = 50 μM) have a temperature of elongation (T e) of ∼45 °C. Adding 0.25 and 0.5 equiv of TFPB does not significantly change the T e values, but leads to lower CD intensities at 20 °C. This observation suggests that TPA-amide monomers assemble into different structures such as chiral polymer stacks and small, disordered aggregates. Due to the low intensity of CD signals, it is unclear whether higher TFPB equivalents cause a change in the T e values. On the other hand, the UV-Vis cooling curves show that the temperature of aggregation of TPA-amide seems to be slightly shifted in the presence of 0.25 and 0.5 equiv of TFPB ([Figure S18b]). This may suggest that TFPB also drives the aggregation of TPA-amide into achiral, small aggregates to some degree.
Further evidence for the nature of the interaction between TPA-amide with TFPB is obtained with infrared spectroscopy ([Figure 7]). A solution of TPA-amide in Dec/DCE (9:1 v/v, 2 mM) shows an NH stretch at 3300 cm−1, indicating the formation of a 3-fold intermolecular H-bonding network, which is in sharp contrast to the free NH peak observed at 3457 cm−1 for TPA-amide in chloroform, where it is molecular dissolved ([Figure S19]).[24] [39] Adding 0.5 equiv TFPB to TPA-amide in Dec/DCE results in two NH stretches (3453 and 3315 cm−1 , yellow line in [Figure 7]). The intensity of the peak at 3315 cm−1 exceeds the free NH peaks, suggesting that TPA-amide can still efficiently form an intermolecular H-bonding network at this TFPB concentration. Further increasing TFPB to 1 equiv increases the proportion of the free NH stretch compared to the bonded NH stretch ([Figure 7], blue line). There is also a clear shift of the bonded-NH peak to a longer wavenumber, which suggests less-ordered H-bonding. These observations corroborate that TFPB can disrupt the H-bonding network of TPA-amide, which could explain low chirality observed by CD spectroscopy. In our attempt to simulate the data with theoretical models (see the Supporting Information) we could not further clarify the role of TFPB with TPA-amide, as we have done in earlier studies.[38] We thus propose that TFPB forms a dimer or small aggregates with TPA-amide through a sequestration mechanism. Although no mechanistic analysis is performed with the TPA-amide9/TFPB system, it is most likely that TFPB can intercalate into the stack of TPA-amide9.

Conclusions
We have investigated the co-assembly of B–N FLP supramolecular systems by varying donor strength and types of homo-interactions in the nitrogen-centered components to obtain stable, long-range ordered B–N systems. Spectroscopic analysis of the electron-rich TPA-alkyne indicates the formation of charge transfer complexes with TFPB in the ground state. On the other hand, the triphenylamines with electron-withdrawing groups (i.e., TPA-ester, TPA-amide and TPA-amide9) could efficiently form charge transfer complexes with TFPB in the excited state, as evidenced in the photoluminescence experiments. In particular, the role of TFPB in the interactions with these electron-poor triphenylamines appears to be directed by their intermolecular homo-interactions. TFPB can reduce the degree of aggregation of TPA-amide polymers by sequestering TPA-amide monomers from the monomer–polymer equilibrium. The conformational flexibility and multimodal H-bonding of the branched side chains of TPA-amide9 increase the tolerance for disorder in the stack upon the intercalation of TFPB. Therefore, both B–N and N–N interactions should be carefully balanced to generate stable B–N supramolecular systems.
Although the goal in generating B–N alternating copolymers has not been achieved, the results highlight the practical potentials of using a commercial boron compound in designing functional supramolecular systems based on B–N FLPs. Further improvements of our current approach with TFPB can focus on designing a new amide-based triphenylamine derivative that incorporates branched side chains such as TPA-amide9 but has weaker homo-interactions. Alternatively, the electron-rich TPA-alkyne can be combined with a synthetic boron compound such as bridged triphenylborane,[40] which is a weaker acceptor than TFPB to create a stable B–N complex. We anticipate that a clever design of B and N components can facilitate the development of stable, efficient B–N supramolecular systems for numerous applications in optoelectronics.
Experimental Section
Materials and Instruments
Anhydrous decalin was purchased from Acros Organics. DCE was purchased from Alfa Aesar. These solvents were purged with argon and stored on molecular sieves for the preparation of stock solutions. TFPB was obtained from Tokyo Chemical Industry. All other solvents were obtained from Biosolve and Aldrich. Other chemicals were obtained from Aldrich and used as received. (S)-(-)-Citronellol was purchased from Aldrich and converted into the corresponding (S)-3,7-dimethyloctanol and (S)-3,7-dimethyloctylamine according to a previously reported procedure.[41]
UV-Vis, CD and photoluminescence measurements were performed on a Jasco J-815 spectropolarimeter, equipped with a Jasco MPTC-490S Peltier temperature-controlled multi-cell holder and a Jasco FMO-427S/15 emission monochromator. Photoluminescence spectra were recorded using a Varian Cary Eclipse fluorescence spectrometer. X-band EPR spectra were acquired on a Bruker EMX X-band spectrometer, equipped with an ER 4112HV-CF100 cryostat and a Hoenle Bluepoint 4 light source. IR spectra were recorded on a Perkin– Elmer spectrum two FTIR spectrometers. CPL spectra were recorded on a homebuilt instrument employing a photoelastic modulator (Hinds) to discriminate between left- and right-handed photons. The apparatus uses multichannel detection involving single photon counting. The sample is excited with filtered light from a Hg lamp incoming under an angle of 90° with respect to the emission collection. To suppress artefacts resulting from linear polarization of the luminescence through photoselection, the excitation light was polarized in the horizontal plane spanned by the directions of the excitation beam and the direction of emission collection. 1H NMR and 13C NMR measurements were carried out on a Varian Gemini 400 MHz (100 MHz for 13C). Proton chemical shifts are reported in ppm downfield from tetramethylsilane (TMS). Carbon chemical shifts are reported using the resonance of CDCl3 as internal standard. MALDI-TOF-MS data were acquired using a PerSeptive Biosystems Voyager-DE PRO spectrometer using α-cyano-4-hydroxycinnamic acid and 2-[(2E)-3-(4-tert-butylphenyl)-2- methylprop-2-enylidene]malononitrile as matrices.
Sample Preparation for Spectroscopic Measurements
To prepare stock solutions, TPA derivatives were dissolved in Dec and TFPB was dissolved in DCE in an N2 equilibrated glovebox. TPA stock solutions were heated up, sonicated, and cooled to room temperature. Stock solutions of TPA and TFPB were then mixed in the cuvettes, and extra solvents were added to adjust the mixing ratio if necessary. All cuvettes were sealed before taking out from the glovebox. Quartz cuvettes with an optical path length of 1 cm, 4 mm or 2 mm were used.
Synthesis of TPA-alkyne
In a Schlenk tube, tris(4-iodophenyl)amine (101.4 mg, 0.16 mmol), 1,2,3-tris(dodecyloxy)-5-ethynylbenzene (349 mg, 0.53 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (1.1 mL, 0.16 mmol) were combined in dry toluene (10 mL) under an argon atmosphere. Then, CuI (25.5 mg, 0.13 mmol) and PdCl2(PPh3)4 (26.0 mg, 0.04 mmol) were added. The mixture was stirred at room temperature for 22 h and diluted with 100 mL CHCl3. The organic layer was washed with 100 mL water, 2 × 100 mL 1M HCl, 100 mL saturated NaHCO3 and 100 mL brine, dried over MgSO4, filtered and evaporated under reduced pressure. The crude product was purified by column chromatography (50 g SiO2) by using heptane/dichloromethane (1:1 v/v) as eluents. The product was obtained as a light-yellow solid (yield = 274 mg, 76%). 1H NMR (CDCl3, 400 MHz): δ = 7.45–7.37 (d, 6 H), 7.09–7.02 (d, 6 H), 6.72 (s, 6 H), 3.97 (m, 18 H), 1.77 (m, 18 H), 1.49–1.26 (m, 162 H), 0.92–0.84 (t, 27H). 13C NMR (CDCl3, 100 MHz): δ = 153.00, 132.68, 123.99, 110.06, 73.55, 69.14, 31.95, 31.93, 30.32, 29.76, 29.71, 29.67, 29.65, 29.61, 29.41, 29.37, 29.35, 26.10, 22.70, 14.12. MALDI-TOF-MS: calc. for C150H243NO9: 2202.86; found: 2202.87.
Synthesis of TPA-amide
In a 50-mL flask, 4,4',4''-nitrilotribenzoic acid (300 mg, 0.80 mmol) and triethylamine (1 mL, 7.2 mmol) were combined in dry DMF (12 mL). The mixture was cooled in an ice bath and purged with argon. In another vial, pentafluorophenyl trifluoroacetate (620 µL, 3.6 mmol) was dissolved in dry DMF (1 mL) and added dropwise to the reaction mixture. The reaction mixture was stirred in an ice bath for 1 h then at room temperature for 3 h. In another vial, (S)-3,7-dimethyloctylamine (422 mg, 2.7 mmol) was dissolved in dry DMF (2 mL) and added to the reaction mixture. The reaction mixture was stirred at room temperature overnight, diluted with 100 mL water and extracted with 2 × 100 mL chloroform. The organic phase was washed with 200 mL 1M HCl, 200 mL 1M NaOH and 200 mL brine, dried over MgSO4, filtered and evaporated under reduced pressure, giving light yellow oil. Diethyl ether was added to the crude product, leading to white precipitates. The mixture was heated, sonicated and cooled to room temperature. The solid was collected by vacuum filtration, washed with diethyl ether and dried, giving an off-white solid. Chloroform was added on top of the filter paper to dissolve the product and collected in a flask. Solvent evaporation gave a white solid (yield = 228 mg, 36%). 1H NMR (CDCl3, 400 MHz): δ = 7.67 (d, 6 H), 7.10 (d, 6 H), 5.96 (s, 3 H), 3.49 (m, 6H), 1.66–1.26 (m, 30 H), 0.95 (d, 9 H), 0.86 (d, 18 H). 13C NMR (CDCl3, 100 MHz): δ = 166.67, 149.24, 129.94, 128.35, 123.88, 39.23, 38.28, 37.15, 36.81, 30.81, 27.95, 24.66, 22.70, 22.60, 19.57. MALDI-TOF-MS: calc. for C51H78N4O3: 794.61; found: 794.63.
Synthesis of Tert-butyl (S)-(1,5-bis(dodecylamino)-1,5-dioxopentan-2-yl)carbamate
In a 500-mL flask, Boc-glutamic acid (4 g, 16.2 mmol), dodecylamine (7.5 g, 40.4 mmol) and 1-hydroxybenzotriazole hydrate (7.65 g, 56.6 mmol) were dissolved in dichloromethane (200 mL). Then, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (10.85 g, 56.6 mmol) and diisopropylethylamine (12.68 mL, 72.8 mmol) were added. The mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure. The crude was diluted with THF (100 mL), poured into 5% Na2CO3 solution (900 mL) and stirred at room temperature. The precipitates were collected by vacuum filtration, washed with water and dried at 80 °C under reduced pressure. A white solid was obtained (9.7 g) and used for the next step without further purification.
Synthesis of (S)-2-Amino-N1,N5 -didodecylpentanediamide
In a 50-mL flask, tert-butyl (S)-(1,5-bis(dodecylamino)-1,5-dioxopentan-2-yl)carbamate (4.3 g) was dissolved in dichloromethane (90 mL), followed by addition of trifluoroacetic acid (12 mL). The mixture was stirred at room temperature overnight. The mixture was evaporated then co-evaporated with toluene, giving light yellow viscous oil. The crude was dissolved in ethanol (50 mL), poured into 1% Na2CO3 solution (800 mL) and stirred at room temperature. The precipitates were collected by vacuum filtration and recrystallized in ethanol (100 mL), giving a white/pink solid. The solid was dissolved in chloroform, passed through filter paper and dried under reduced pressure. A white solid was obtained (2.7 g, 76%). 1H NMR (CDCl3, 400 MHz): δ = 7.31 (t, 1 H), 6.08 (t, 1 H), 3.41 (t, 1 H), 3.23 (m, 4 H), 2.38–2.25 (m, 2 H), 1.94 (q, 2 H), 1.51–1.26 (m, 40 H), 0.88 (t, 6 H). 13C NMR (CDCl3, 100 MHz): δ = 174.69, 174.62, 172.60, 172.52, 54.06, 39.62, 39.48, 39.08, 38.96, 33.28, 33.23, 31.92, 29.66, 29.64, 29.60, 29.56, 29.36, 29.30, 26.97, 22.69, 14.13. MALDI-TOF-MS: calc. for C29H59N3O2: 481.46; found: 341.33 and 504.45.
Synthesis of TPA-amide9
In a 50-mL flask, 4,4',4''-nitrilotribenzoic acid (252 mg, 0.67 mmol), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (746 mg, 2.32 mmol) and dry DMF (10 mL) were combined and purged with N2. Diisopropylethylamine (810 µL, 4.65 mmol) was added and the solution was stirred at room temperature for 20 min. In another vial, (S)-2-amino-N 1,N 5-didodecylpentanediamide (1.1 g, 2.28 mmol) was dissolved in hot DMF (5 mL) and then added into the reaction mixture. The reaction mixture was stirred at 100 °C for 5 days. After cooling to room temperature, the mixture was diluted with chloroform (150 mL) and washed with water, 1M HCl, 1M NaOH and brine (with trifluoracetic acid added). The organic layer was collected, dried over MgSO4, filtered and evaporated under reduced pressure to give yellow solids. The crude product was dispersed in hot acetonitrile, and insoluble materials were collected by vacuum filtration, giving a creamy solid. The solid was subjected to another re-crystallization. A white solid was collected and dried at 80 °C under reduced pressure (922 mg, 79%). 1H NMR (CDCl3/CF3COOD, 400 MHz): δ = 7.76 (d, 6 H), 7.16 (d, 6 H), 3.34 (m, 12 H), 2.70 (m, 6 H), 2.31 (m, 6 H), 1.56–1.03 (m, 120 H), 0.88 (t, 18 H). 13C NMR (CDCl3/CF3COOD, 100 MHz): δ = 176.39, 172.94, 170.68, 151.01, 129.81, 126.53, 124.74, 53.90, 42.26, 41.47, 32.28, 31.60, 29.97, 29.96, 29.89, 29.87, 29.76, 29.73, 29.70, 29.38, 28.79, 28.56, 27.00, 26.98, 23.01, 14.18. MALDI-TOF-MS: calc. for C108H186N10O9: 1797.44; found: 1790.45.
Synthesis of TPA-ester
A flask equipped with an argon outlet was charged with 4,4',4''-nitrilotribenzoic acid (142.9 mg, 0.379 mmol) and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (427.2 mg, 1.331 mmol), and dry DMF (7.6 mL) was added. The mixture was stirred under an argon atmosphere and 1,8-diazabicyclo[5.4.0]undec-7-ene (0.4 mL) was added. After stirring for 25 min at room temperature, (S)-3,7-dimethyloctanol (221.6 mg, 1.400 mmol) was added with dry DMF (5 mL). The mixture was left to stir overnight. Afterwards, it was extracted with 100 mL CHCl3, 3 × 100 mL H2O, 100 mL 1M HCl, 100 mL H2O and 100 mL brine. Afterwards, it was dried with MgSO4 and the solvent was evaporated. Column chromatography with the eluent (heptane/CHCl3 1:1 v/v) and evaporation of solvent resulted in TPA-ester as a colorless liquid (95.6 mg, 32%). 1H NMR (CDCl3, 400 MHz): δ = 7.98–7.91 (d, 6 H), 7.16–7.09 (d, 6 H), 4.39–4.30 (m, 6 H), 1.611.11 (m, 30 H), 0.95 (d, 9 H), 0.86 (d, 18 H). 13C NMR (CDCl3, 100 MHz): δ = 166.03, 150.32, 131.14, 125.83, 123.77, 63.56, 39.20, 37.16, 35.62, 30.00, 27.95, 24.64, 22.65, 19.63. MALDI-TOF-MS: calc. for C51H71NO6: 797.56; found: 797.56.
Additional characterization data, details of theoretical modeling and [Figures S1]–[S21] are available in the Supporting Information.
Funding Information
The work received funding from The Netherlands Organization for Scientific Research (NWO-TOP PUNT Grant No. 10018944) and the Dutch Ministry of Education, Culture and Science (Gravitation Program 024.001.035).
No conflict of interest has been declared by the author(s).
Acknowledgment
The authors would like to thank Beatrice Adelizzi for providing TPA-amide molecules and Martin van Son for his support in film measurements. Professor Shigehiro Yamaguchi (Nagoya University, Japan) is acknowledged for stimulating discussions on boron–nitrogen interactions.
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0041-1727235.
This paper is dedicated to the 65th anniversary of Peter Bäuerle to honor his seminal contributions to the field of organic electronic materials.
-
References
- 1 Aida T, Meijer EW. Isr. J. Chem. 2020; 60: 33
- 2 Hashim PK, Bergueiro J, Meijer EW, Aida T. Prog. Polym. Sci. 2020; 105: 101250
- 3 Adelizzi B, Van Zee NJ, de Windt LN. J, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2019; 141: 6110
- 4 Mabesoone MF. J, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2020; 142: 19781
- 5 Helmich F, Lee CC, Nieuwenhuizen MM. L, Gielen JC, Christianen PC. M, Larsen A, Fytas G, Leclère PE. L. G, Schenning AP. H. J, Meijer EW. Angew. Chem. Int. Ed. 2010; 49: 3939
- 6 Rao KV, Miyajima D, Nihonyanagi A, Aida T. Nat. Chem. 2017; 9: 1133
- 7 Sarkar A, Sasmal R, Empereur-Mot C, Bochicchio D, Kompella SV. K, Sharma K, Dhiman S, Sundaram B, Agasti SS, Pavan GM, George SJ. J. Am. Chem. Soc. 2020; 142: 7606
- 8 Das A, Ghosh S. Angew. Chem. Int. Ed. 2014; 53: 2038
- 9 Haid S, Marszalek M, Mishra A, Wielopolski M, Teuscher J, Moser JE, Humphry-Baker R, Zakeeruddin SM, Grätzel M, Bäuerle P. Adv. Funct. Mater. 2012; 22: 1291
- 10 Gon M, Tanaka K, Chujo Y. Bull. Chem. Soc. Jpn. 2019; 92: 7
- 11 Mellerup SK, Wang S. Trends Chem. 2019; 1: 77
- 12 Wakamiya A, Yamaguchi S. Bull. Chem. Soc. Jpn. 2015; 88: 1357
- 13 Matsuo K, Saito S, Yamaguchi S. J. Am. Chem. Soc. 2014; 136: 12580
- 14 Willms A, Schumacher H, Tabassum T, Qi L, Scott SL, Hausoul PJ. C, Rose M. ChemCatChem 2018; 10: 1835
- 15 Goushi K, Yoshida K, Sato K, Adachi C. Nat. Photonics 2012; 6: 253
- 16 Ye T, Chen W, Jin S, Hao S, Zhang X, Liu H, He D. ACS Appl. Mater. Interfaces 2019; 11: 14004
- 17 Wang M, Nudelman F, Matthes RR, Shaver MP. J. Am. Chem. Soc. 2017; 139: 14232
- 18 Chen L, Liu R, Yan Q. Angew. Chem. Int. Ed. 2018; 57: 9336
- 19 Adelizzi B, Chidchob P, Tanaka N, Lamers BA. G, Meskers SC. J, Ogi S, Palmans AR. A, Yamaguchi S, Meijer EW. J. Am. Chem. Soc. 2020; 142: 16681
- 20 Yokoyama D. J. Mater. Chem. 2011; 21: 19187
- 21 Weck M, Dunn AR, Matsumoto K, Coates GW, Lobkovsky EB, Grubbs RH. Angew. Chem. Int. Ed. 1999; 38: 2741
- 22 Hsu SM, Lin YC, Chang JW, Liu YH, Lin HC. Angew. Chem. Int. Ed. 2014; 53: 1921
- 23 Kivala M, Pisula W, Wang S, Mavrinskiy A, Gisselbrecht JP, Feng X, Müllen K. Chem. Eur. J. 2013; 19: 8117
- 24 Adelizzi B, Filot IA. W, Palmans AR. A, Meijer EW. Chem. Eur. J. 2017; 23: 6103
- 25 Yang D, Duan P, Zhang L, Liu M. Nat. Commun. 2017; 8: 1
- 26 Chen JY, Kadam G, Gupta A. Chem. Eur. J. 2018; 24: 14668
- 27 Miao W, Qin L, Yang D, Jin X, Liu M. Chem. Eur. J. 2015; 21: 1064
- 28 Holtrop F, Jupp AR, van Leest NP, Paradiz Dominguez M, Williams RM, Brouwer AM, de Bruin B, Ehlers AW, Slootweg JC. Chem. Eur. J. 2020; 26: 9005
- 29 Roessler MM, Salvadori E. Chem. Soc. Rev. 2018; 47: 2534
- 30 Amthor S, Noller B, Lambert C. Chem. Phys. 2005; 316: 141
- 31 Quinton C, Alain-Rizzo V, Dumas-Verdes C, Miomandre F, Clavier G, Audebert P. RSC Adv. 2014; 4: 34332
- 32 Kwaan RJ, Harlan CJ, Norton JR. Organometallics 2001; 20: 3818
- 33 Lawrence EJ, Oganesyan VS, Wildgoose GG, Ashley AE. Dalton Trans. 2013; 42: 782
- 34 Li A, Chu N, Liu J, Liu H, Wang J, Xu S, Cui H, Zhang H, Xu W, Ma Z. Mater. Chem. Front. 2019; 3: 2768
- 35 Mamada M, Tian G, Nakanotani H, Su J, Adachi C. Angew. Chem. Int. Ed. 2018; 57: 12380
- 36 Nakanotani H, Furukawa T, Morimoto K, Adachi C. Sci. Adv. 2016; 2: e1501470
- 37 Sánchez-Carnerero EM, Agarrabeitia AR, Moreno F, Maroto BL, Muller G, Ortiz MJ, de la Moya S. Chem. Eur. J. 2015; 21: 13488
- 38 Weyandt E, Mabesoone MF. J, de Windt LN. J, Meijer EW, Palmans AR. A, Vantomme G. Org. Mater. 2020; 02: 129
- 39 Stals PJ. M, Everts JC, de Bruijn R, Filot IA. W, Smulders MM. J, Martín-Rapún R, Pidko EA, de Greef TF. A, Palmans AR. A, Meijer EW. Chem. Eur. J. 2010; 16: 810
- 40 Kushida T, Shuto A, Yoshio M, Kato T, Yamaguchi S. Angew. Chem. Int. Ed. 2015; 54: 6922
- 41 Terashima T, Mes T, De Greef TF. A, Gillissen MA. J, Besenius P, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2011; 133: 4742
Publication History
Received: 28 February 2021
Accepted: 16 March 2021
Article published online:
01 April 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Aida T, Meijer EW. Isr. J. Chem. 2020; 60: 33
- 2 Hashim PK, Bergueiro J, Meijer EW, Aida T. Prog. Polym. Sci. 2020; 105: 101250
- 3 Adelizzi B, Van Zee NJ, de Windt LN. J, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2019; 141: 6110
- 4 Mabesoone MF. J, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2020; 142: 19781
- 5 Helmich F, Lee CC, Nieuwenhuizen MM. L, Gielen JC, Christianen PC. M, Larsen A, Fytas G, Leclère PE. L. G, Schenning AP. H. J, Meijer EW. Angew. Chem. Int. Ed. 2010; 49: 3939
- 6 Rao KV, Miyajima D, Nihonyanagi A, Aida T. Nat. Chem. 2017; 9: 1133
- 7 Sarkar A, Sasmal R, Empereur-Mot C, Bochicchio D, Kompella SV. K, Sharma K, Dhiman S, Sundaram B, Agasti SS, Pavan GM, George SJ. J. Am. Chem. Soc. 2020; 142: 7606
- 8 Das A, Ghosh S. Angew. Chem. Int. Ed. 2014; 53: 2038
- 9 Haid S, Marszalek M, Mishra A, Wielopolski M, Teuscher J, Moser JE, Humphry-Baker R, Zakeeruddin SM, Grätzel M, Bäuerle P. Adv. Funct. Mater. 2012; 22: 1291
- 10 Gon M, Tanaka K, Chujo Y. Bull. Chem. Soc. Jpn. 2019; 92: 7
- 11 Mellerup SK, Wang S. Trends Chem. 2019; 1: 77
- 12 Wakamiya A, Yamaguchi S. Bull. Chem. Soc. Jpn. 2015; 88: 1357
- 13 Matsuo K, Saito S, Yamaguchi S. J. Am. Chem. Soc. 2014; 136: 12580
- 14 Willms A, Schumacher H, Tabassum T, Qi L, Scott SL, Hausoul PJ. C, Rose M. ChemCatChem 2018; 10: 1835
- 15 Goushi K, Yoshida K, Sato K, Adachi C. Nat. Photonics 2012; 6: 253
- 16 Ye T, Chen W, Jin S, Hao S, Zhang X, Liu H, He D. ACS Appl. Mater. Interfaces 2019; 11: 14004
- 17 Wang M, Nudelman F, Matthes RR, Shaver MP. J. Am. Chem. Soc. 2017; 139: 14232
- 18 Chen L, Liu R, Yan Q. Angew. Chem. Int. Ed. 2018; 57: 9336
- 19 Adelizzi B, Chidchob P, Tanaka N, Lamers BA. G, Meskers SC. J, Ogi S, Palmans AR. A, Yamaguchi S, Meijer EW. J. Am. Chem. Soc. 2020; 142: 16681
- 20 Yokoyama D. J. Mater. Chem. 2011; 21: 19187
- 21 Weck M, Dunn AR, Matsumoto K, Coates GW, Lobkovsky EB, Grubbs RH. Angew. Chem. Int. Ed. 1999; 38: 2741
- 22 Hsu SM, Lin YC, Chang JW, Liu YH, Lin HC. Angew. Chem. Int. Ed. 2014; 53: 1921
- 23 Kivala M, Pisula W, Wang S, Mavrinskiy A, Gisselbrecht JP, Feng X, Müllen K. Chem. Eur. J. 2013; 19: 8117
- 24 Adelizzi B, Filot IA. W, Palmans AR. A, Meijer EW. Chem. Eur. J. 2017; 23: 6103
- 25 Yang D, Duan P, Zhang L, Liu M. Nat. Commun. 2017; 8: 1
- 26 Chen JY, Kadam G, Gupta A. Chem. Eur. J. 2018; 24: 14668
- 27 Miao W, Qin L, Yang D, Jin X, Liu M. Chem. Eur. J. 2015; 21: 1064
- 28 Holtrop F, Jupp AR, van Leest NP, Paradiz Dominguez M, Williams RM, Brouwer AM, de Bruin B, Ehlers AW, Slootweg JC. Chem. Eur. J. 2020; 26: 9005
- 29 Roessler MM, Salvadori E. Chem. Soc. Rev. 2018; 47: 2534
- 30 Amthor S, Noller B, Lambert C. Chem. Phys. 2005; 316: 141
- 31 Quinton C, Alain-Rizzo V, Dumas-Verdes C, Miomandre F, Clavier G, Audebert P. RSC Adv. 2014; 4: 34332
- 32 Kwaan RJ, Harlan CJ, Norton JR. Organometallics 2001; 20: 3818
- 33 Lawrence EJ, Oganesyan VS, Wildgoose GG, Ashley AE. Dalton Trans. 2013; 42: 782
- 34 Li A, Chu N, Liu J, Liu H, Wang J, Xu S, Cui H, Zhang H, Xu W, Ma Z. Mater. Chem. Front. 2019; 3: 2768
- 35 Mamada M, Tian G, Nakanotani H, Su J, Adachi C. Angew. Chem. Int. Ed. 2018; 57: 12380
- 36 Nakanotani H, Furukawa T, Morimoto K, Adachi C. Sci. Adv. 2016; 2: e1501470
- 37 Sánchez-Carnerero EM, Agarrabeitia AR, Moreno F, Maroto BL, Muller G, Ortiz MJ, de la Moya S. Chem. Eur. J. 2015; 21: 13488
- 38 Weyandt E, Mabesoone MF. J, de Windt LN. J, Meijer EW, Palmans AR. A, Vantomme G. Org. Mater. 2020; 02: 129
- 39 Stals PJ. M, Everts JC, de Bruijn R, Filot IA. W, Smulders MM. J, Martín-Rapún R, Pidko EA, de Greef TF. A, Palmans AR. A, Meijer EW. Chem. Eur. J. 2010; 16: 810
- 40 Kushida T, Shuto A, Yoshio M, Kato T, Yamaguchi S. Angew. Chem. Int. Ed. 2015; 54: 6922
- 41 Terashima T, Mes T, De Greef TF. A, Gillissen MA. J, Besenius P, Palmans AR. A, Meijer EW. J. Am. Chem. Soc. 2011; 133: 4742