

Subscribe to RSS
DOI: 10.1055/s-0034-1371528
Pulmonary Manifestations of Polymyositis/Dermatomyositis
Authors
Address for correspondence
Publication History
Publication Date:
25 March 2014 (online)
Abstract
The idiopathic inflammatory myopathies are a group of connective tissue diseases marked by varying degrees of muscle inflammation and clinical involvement of multiple organs, most notably, the lung. Pulmonary manifestations consist primarily of interstitial lung disease (ILD), which is associated with significant morbidity and mortality in myositis patients. Several myositis-specific antibodies have been discovered, as well as antibodies targeting various aminoacyl-tRNA synthetase enzymes. These antibodies are associated with various clinical features and a risk for developing ILD, and their presence carries a prognostic value in myositis patients. Steroids remain the first-line treatment for myositis-associated ILD and the antisynthetase syndrome, though other traditional immunosuppressants have demonstrated efficacy in numerous studies. While a majority of patients experience either stabilization or improvement in lung imaging and function, fatal progression is still reported in a significant number of cases. Further research is needed to develop more effective and targeted therapies.
Keywords
idiopathic inflammatory myopathies - polymyositis - dermatomyositis - interstitial lung disease - nonspecific interstitial pneumonia - myositis-specific autoantibodiesThe Idiopathic Inflammatory Myopathies
The idiopathic inflammatory myopathies (IIMs) are a group of relatively rare connective tissue diseases marked by varying degrees of muscle inflammation. The main forms of adult IIM include polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM). Amyopathic DM (ADM) contains the classic skin findings of DM in the absence of overt muscle involvement.[1] With the exception of IBM, each of these entities can present with a wide range of clinical findings involving multiple organ systems, including the heart, skin, joints, and lung.
Pulmonary involvement in DM/PM is associated with worse outcomes and increased mortality[2] [3] and is the primary reason for hospital admission in a majority of patients.[4] Depending on the methods used to detect interstitial lung disease (ILD), its prevalence in patients with DM/PM ranges from 19.9 to 78%,[2] [5] [6] [7] [8] [9] and its presentation can range from fulminant to subclinical.[10] [11] Nevertheless, ILD is not one of the original criteria used by Bohan and Peter to diagnose DM or PM.[12] [13] Although it most frequently develops during or after the appearance of muscle inflammation, ILD precedes the diagnosis of DM/PM in 13 to 37.5% of patients[2] [4] [14] [15] and tends to be associated with higher rates of fever and joint involvement.[7]
The Antisynthetase Syndrome
In recent years, several myositis-specific autoantibodies (MSAs) have been discovered: antisignal recognition particle (anti-SRP) antibodies, anti-Mi-2 antibodies, anti-PM/Scl antibodies, anti-aminoacyl-tRNA synthetase (ARS) antibodies, anticlinically ADM-140-kDa polypeptide antibodies (anti-CADM-140 antibodies or melanoma differentiation–associated gene 5 [MDA-5]), and anti-155/140-kDa polypeptide antibodies (anti-155/140 antibodies)[16] [17] [18] [19] ([Table 1]). Among these MSAs, ARS antibodies target the cytoplasmic ARS enzymes that are responsible for loading each tRNA with a specific amino acid before it is incorporated into a growing peptide at the level of the ribosome.[20] There are 20 different amino acids, each of which has a cognate tRNA synthetase.[21] To date, antibodies specific for eight of these synthetases have been detected and implicated in connective tissue disease: antihistidyl- (anti-Jo-1), antithreonyl- (anti-PL-7), antialanyl- (anti-PL-12), antiisoleucyl- (anti-OJ), antiasparaginyl- (anti-KS), antiglycyl- (anti-EJ), antiphenylalanyl- (anti-Zo), and anti-yrosyl-tRNA- (anti-YRS) tRNA synthetase[22] [23] [24] ([Table 2]).
|
Myositis-associated antibodies |
Target antigen |
Prevalence in IIM[17] [18] [19] [27] [37] [38] [39] [44] [48] [93] |
Clinical features[17] [27] [37] [38] [39] [42] [54] [55] [56] [57] [59] |
|---|---|---|---|
|
Anti-Ro52 |
Extractable nuclear antigen Ro52 |
13–26% |
Associated with more severe ILD |
|
Anti-MDA-5 |
MDA-5 RNA helicase |
20–25%[a] |
Skin ulceration; CADM; rapidly progressing/acute interstitial pneumonitis |
|
Anti-155/140 |
155/140-kDa polypeptides |
7–16% |
Highest risk of malignancy, lower risk of ILD |
|
Anti-SRP |
Cytoplasmic signal recognition particle |
5–6% |
Severe myopathy and weakness; risk of ILD is not increased |
|
Anti-Mi-2 |
Nuclear helicase protein |
14% |
Milder myopathy and classic DM findings; risk of ILD is not increased |
Abbreviations: CADM, clinically amyopathic dermatomyositis; DM, dermatomyositis; IIM, idiopathic inflammatory myopathy; ILD, interstitial lung disease; MDA-5, melanoma differentiation–associated gene 5; SRP, signal recognition particle.
a Prevalence is reported in patients with DM/CADM only, as anti-MDA-5 antibodies are rarely found in PM.
|
Antisynthetase antibodies (ARS) |
Target antigen |
% of ARS antibodies detected[14] [20] [23] [26] [31] [50] [79] [93] |
||
|---|---|---|---|---|
|
Anti-Jo-1 |
Histidyl-tRNA synthetase |
8–18% |
36–88% |
Classic DM, ILD |
|
Anti-EJ |
Glycyl-tRNA synthetase |
5–10% |
7–23% |
Classic DM and CADM, ILD |
|
Anti-PL-7 |
Threonyl-tRNA synthetase |
5% |
9–25% |
Classic DM, ILD |
|
Anti-OJ |
Isoleucyl-tRNA synthetase |
3% |
5–8% |
Isolated ILD |
|
Anti-PL-12 |
Alanyl-tRNA synthetase |
1% |
2–11% |
Isolated ILD, CADM |
|
Anti-KS |
Asparaginyl-tRNA synthetase |
1% |
4–8% |
Isolated ILD |
|
Anti-Zo |
Phenylalanyl-tRNA synthetase |
< 1% |
< 1% |
|
|
Anti-YRS |
Tyrosyl-tRNA synthetase |
< 1% |
< 1% |
Abbreviations: ARS, antiaminoacyl-tRNA synthetase; CADM, clinically amyopathic dermatomyositis; DM, dermatomyositis; IIM, idiopathic inflammatory myopathy; ILD, interstitial lung disease.
The presence of an ARS antibody in conjunction with inflammatory myositis or ILD has collectively come to be known as the antisynthetase syndrome,[25] with additional clinical features that can include nonerosive arthritis, fever, mechanic's hands ([Fig. 1]), and Raynaud phenomenon.[26] [27] [28] While the concomitant presence of other autoimmune antibodies is not uncommon, overlap between the ARS antibodies themselves is rare.[14] [20] [27] [28] Specific ARS antibodies are associated with different subphenotypes characterized by a varying prevalence of myositis, ILD, and other clinical manifestations of connective tissue disease.[27] In general, the presence of any ARS antibody confers a substantially higher risk for developing ILD, with multiple studies demonstrating a prevalence of 67 to 100% depending on the specific antibody identified.[20] [25] [27] [29] [30]
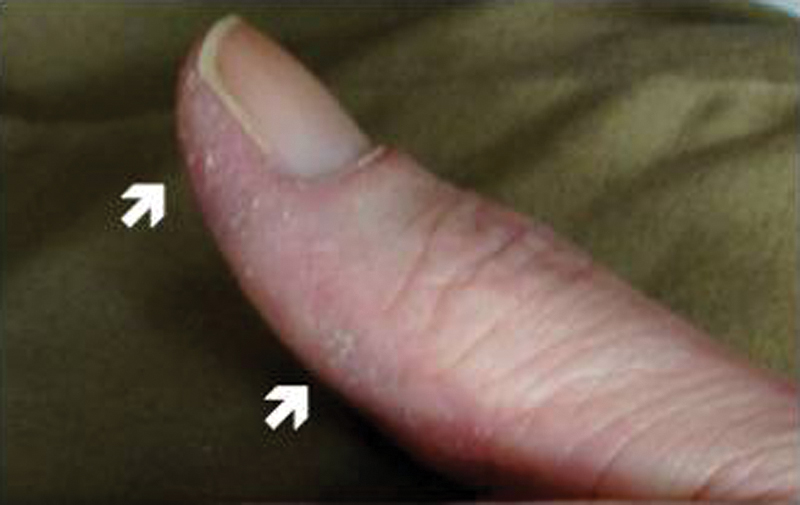
Of the eight known ARS antibodies, anti-Jo-1 is the most common, being present in 15 to 30% of patients with IIM.[18] [31] Clinically evident myositis occurs in greater than 50% of patients with the anti-Jo-1 antibody,[27] and ILD is also extremely common, occurring in 67 to 86% of cases.[9] [15] [26] [29] [32] [33]
Collectively, anti-PL-7, anti-PL-12, anti-KS, and anti-OJ confer the greatest risk of developing ILD,[20] [21] [22] [27] [29] [34] [35] and patients with these antibodies are also more likely to have ILD either in isolation or preceding the diagnosis of DM/PM. For example, one study demonstrated that the rates of ILD existing in the absence of clinically evident myositis were 33, 63, and 77% in patients positive for anti-PL-12, anti-KS, and anti-OJ, respectively.[27] In another study, 35% of patients with anti-PL-7 antibodies had ILD that preceded the diagnosis of antisynthetase sydndrome.[34]
Anti-MDA-5 (Anti-CADM-140) Antibodies
In addition to the ARS antibodies, a 140-kDa cytoplasmic protein was identified as being highly associated with clinically ADM (C-ADM) and subsequently named anti-CADM-140.[16] [36] [37] In 2009, Sato et al discovered that anti-CADM-140 reacts with an RNA helicase encoded by MDA-5, and the antibody was subsequently renamed as anti-MDA-5.[36] Despite its original name, anti-MDA-5 antibodies can be associated with clinically active myositis. Anti-MDA-5 often produces a phenotype that mimics the antisynthetase syndrome[28] [37] [38]; however, it is also associated with characteristic skin findings including tender palmar papules and deep ulcerations with punched out borders and necrosis[28] [38] [39] ([Fig. 2]). Furthermore, anti-MDA-5 antibodies are rarely found in other connective tissue diseases such as rheumatoid arthritis and systemic lupus erythematosus.[37]

Anti-MDA-5 antibodies confer a high risk for the development of ILD and have been associated with rapidly progressive and acute/subacute interstitial pneumonia in several cohorts.[16] [37] [38] [39] [40] [41] [42] [43] [44] [45] Furthermore, the presence of anti-MDA-5 antibodies represents an individual risk factor for death from ILD in DM patients.[37] [41] [42] [44] In one study, anti-MDA-5 antibody levels correlated with the severity of high-resolution computed tomography (CT) scores in patients with ILD. In addition, patients with anti-MDA-5 levels greater than 500 units/mL were found to have more treatment-resistant lung disease and skin ulcerations, with a higher rate of respiratory failure and death compared with patients with levels less than 500 units/mL.[38] Overall, then, anti-MDA-5 titers seem to correlate with treatment response and survival in patients with ILD.[43] [46] [47]
The pathophysiology linking anti-MDA-5 titers with the severity of ILD is poorly understood, though some have suggested a relationship to underlying vascular pathology.[38] [39] MDA-5 is an RNA helicase that recognizes the double stranded RNA of picornaviruses to induce the production of type-1 interferons and tumor necrosis factor as part of the innate antiviral immune response.[37] [43] One hypothesis postulates that anti-MDA-5 antibodies are somehow generated during a viral infection,[36] perhaps through an aberrant, overly exuberant immune response.
Anti-155/140 Antibodies
The third group of myositis-specific antibodies includes those targeting 155/140-kDa polypeptides (anti-155/140 antibodies) corresponding to TIF1 transcription factors. Anti-155/140 antibodies are found in 13 to 16% of DM/PM patients,[17] [40] [45] and they are highly associated with the presence of malignancy in adults.[17] [40] However, in contrast to ARS and anti-MDA-5 antibodies, anti-155/140 antibodies are associated with a lower risk of ILD.[17] [42]
Anti-PM/Scl and Anti-Ro Antibodies
Other antibodies associated with DM/PM and ILD include anti-PM/Scl and anti-Ro52. When present in myositis patients, anti-PM/Scl antibodies are associated with clinical features resembling those of the antisynthetase syndrome.[48] [49] [50] ILD is present with a high frequency in anti-PM/Scl positive patients, though pulmonary disease tends to occur after the appearance of other connective tissue disease manifestations.[50]
Anti-Ro-52 is an antibody found in numerous connective tissue diseases—including systemic lupus erythematosus, mixed connective tissue disease, and DM/PM—and its presence is associated with an increased risk of ILD.[51] Anti-Ro-52 is commonly found in patients with the antisynthetase syndrome[52] and has been reported in 19% of patients with ARS antibodies[27] as well as patients with other myositis-specific antibodies. Overall, 20% of patients with DM/PM express anti-Ro-52 antibodies,[52] [53] though the frequency of anti-Ro-52 is higher among those with ILD; in fact, one cohort reported that 90% of DM/PM patients with ILD were reactive to anti-Ro52.[51] Furthermore, in a cohort of anti-Jo1 positive patients, the presence of anti-Ro52 antibodies was associated with higher total high-resolution computed tomography (HRCT) scores[54] as well as worse myositis, joint involvement, and survival.[32] Collectively, these observations suggest that the coexistence of anti-Ro52 and anti-Jo-1 antibodies may be predictive of a more aggressive disease course.
Anti-SRP and Anti-Mi-2 Antibodies
Anti-SRP antibodies recognize the SRP, a cytoplasmic protein that regulates the movement of proteins through the endoplasmic reticulum.[55] [56] These autoantibodies are found in roughly 5% of IIM patients[18] [19] and are associated with severe myositis marked by profound weakness and highly elevated muscle enzymes. Unlike antisynthetase antibodies, anti-SRP antibodies do not confer an increased risk of developing ILD, and some authors even contend that the risk of pulmonary disease is actually decreased in anti-SRP-positive patients when compared with myositis controls.[55] [56] [57] [58]
Mi-2 is a nuclear helicase protein comprising the nucleosome-remodeling deacetylase complex involved in DNA transcription. Unlike anti-SRP antibodies, anti-Mi-2 antibodies are more common in patients with DM than PM and are often associated with less severe muscle involvement. However, like anti-SRP antibodies, they are not associated with an increased risk of myositis-associated ILD.[55] [59] [60]
Genetic Factors
There is growing evidence that genetic factors may influence the development of ILD in patients with DM/PM and the antisynthetase syndrome. In one cohort of British patients with DM/PM, ILD was more common in patients of black ethnicity.[9] In addition, numerous reports have indicated a higher prevalence of rapidly progressive ILD in cohorts from Eastern Asia with ADM and anti-MDA-5 antibodies,[9] [10] [30] with many cases that have proven refractory to immunosuppressive therapy.[10] However, in an American cohort of anti-MDA-5 positive patients, most cases of ILD were responsive to immunosuppressive therapy.[28] While all of these studies are limited by small sample size, they may indicate that various autoimmune antibodies can be associated with different phenotypes in different ethnic backgrounds.
Genetic studies have identified an association between the DRB1*03-DQA1*05-DQB1*02 haplotype and the presence of both ARS antibodies and ILD. Conversely, HLADRB1*07-DQA1*02-DQB1*02 is associated with a decreased risk of ILD.[61] Furthermore, while HLA-DRB1*0301 is relatively common in Caucasians with ARS antibody-positive DM/PM, the frequency of HLA-DRB1*0301 is low in the Japanese population. However, HLA-DRB1*0405, found in 20% of the Japanese population, is highly associated with anti-MDA-5 antibody–positive DM and associated ILD.[30]
CT Imaging in DM/PM and the Antisynthetase Syndrome
The CT findings of patients with DM/PM and the antisynthetase syndrome can be highly variable depending on the severity and acuity of their pulmonary symptoms. While patients presenting with acute ILD tend to have ground glass opacities and consolidations, patients presenting with a more chronic course of ILD have more evidence of reticulation and honeycombing.[4] In both groups, parenchymal abnormalities are predominantly in a basilar and subpleural distribution.[4] [14] [21] Interestingly, ground glass and reticular opacities can be associated with fatal cases of ILD, while a consolidative pattern is associated with nonfatal disease.[5] [62] [63] Other patterns described with less frequency in DM/PM and the antisynthetase syndrome include traction bronchiectasis and subpleural condensations[35] [64] ([Fig. 3]).

Pulmonary Function Testing
Patients with ILD associated with myositis and the antisynthetase syndrome tend to have a restrictive pattern on pulmonary function tests (PFTs) with generally mild reductions in forced vital capacity (FVC, 70–79% predicted) and mild to moderate reductions in the diffusing capacity for carbon monoxide (DLCO); however, a wide range has been reported, and there have been no definitive studies to suggest that specific myositis-associated antibodies are linked with more severe PFT abnormalities.[4] [8] [9] [15] [24] [37] [64] [65] [66] [67] [68] Total lung capacity (TLC) and FVC have been used to monitor disease progress as well as therapeutic response,[4] [8] [15] [69] and there is evidence that a lower FVC and DLCO at the time of ILD diagnosis predict a worse prognosis.[67] [69] However, PFTs in patients with myositis must be interpreted with caution, since respiratory muscle weakness alone can result in restrictive physiology, even in the absence of ILD. Conversely, PFTs may appear to improve despite persistent or worsening pulmonary disease as muscle weakness responds to corticosteroid or immunomodulatory therapy.[69]
Pneumomediastinum
Spontaneous pneumomediastinum is a rare complication of DM associated with significant morbidity and mortality. One review reported that one-quarter of patients died in the 1st month, with a cumulative survival rate of only 64% at 1 year and 55% at 2 years.[70] Although pneumomediastinum in patients with PM has been reported, the vast majority of cases have occurred in patients with DM.[70] [71] [72] [73] [74] [75] [76] The pathogenesis of pneumomediastinum in patients with myositis is poorly understood, though it is speculated to result from the rupture of subpleural blebs that occur with ILD, leading to dissection of air around perivascular sheaths and into the mediastinum.[75] However, there have been reports of pneumomediastinum occurring in DM patients without known ILD,[71] and some have speculated that vasculopathy may also be playing a role by disrupting the bronchial mucosal barrier and causing subpleural infarctions.[71] [75]
Pathology
Open lung biopsy is often not performed in patients with DM/PM or the antisynthetase syndrome, partly because the results of such a biopsy may not influence the choice of immunosuppressant therapy. However, multiple studies have reported limited pathology with mixed results. Marie et al studied a cohort of 107 patients with DM/PM and found nonspecific interstitial pneumonia (NSIP) in 61%, cryptogenic organizing pneumonia (COP) in 22%, and usual interstitial pneumonia (UIP) in 17% of the 41 biopsy samples available. The prevalence of various MSAs was not evaluated in this study,[67] though there is some evidence that various ARS antibodies may not necessarily be associated with any particular histologic pattern of ILD. For instance, Koreeda et al reported biopsies on four patients positive for different ARS antibodies, each of which demonstrated an NSIP pattern with lymphocytes and plasma cells infiltrating the interstitium.[14] Furthermore, biopsies from three patients with idiopathic interstitial pneumonia in the setting of anti-OJ antibody reactivity demonstrated three different histologic patterns: NSIP, UIP, and COP.[77] In another study, three out of five biopsies in patients with anti-PL-7 antibodies demonstrated a UIP pattern,[34] while NSIP was the most common pattern in a group of 22 patients with anti-Jo-1 antibodies.[15] Further demonstrating the high degree of variability in these studies, Richards et al showed that UIP was three times more common than NSIP in another cohort of anti-Jo-1 positive patients.[33] Moreover, an additional study reported that four out of five patients with myositis who developed respiratory failure were found to have diffuse alveolar damage on biopsy with a mortality rate of 100%.[10] Therefore, while it is possible that associations exist between specific antibodies and certain histologic patterns, no studies to date have been large enough to conclusively determine the nature of such associations.
Treatment
Steroids remain the first-line therapy for ILD in patients with DM/PM or the antisynthetase syndrome.[14] [65] [67] [69] [78] Oral regimens include 1 mg/kg of prednisone daily, often following an initial pulse of intravenous methylprednisolone at a dose of 1,000 mg daily for 3 days in severe cases.[31] While some patients with ILD and myositis will have an adequate response to corticosteroid monotherapy,[31] [65] [78] [79] the need for additional immunosuppression,[14] [65] especially in the case of rapidly progressing acute/subacute ILD,[10] [31] [44] [79] [80] is common. Interestingly, the presence of ARS antibodies may predict a better ILD response to steroids in myositis patients, though additional factors influencing the outcome of steroid treatment include the relative timing of ILD and myositis onset ([Table 3]).[78]
|
Study lead author, year |
Total no. of ILD patients |
CS alone |
CYC |
AZA |
MMF |
CsA |
Tacro |
RTX |
MTX |
IVIG |
Pulmonary improvement, stabilization (%) |
Mortality, n (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Kono et al, 2000[71] |
4 |
2 |
1 |
2 |
NR |
1 (25) |
||||||
|
Lee et al, 2002[10] |
5 |
3 |
3 |
3 |
NR |
5 (100) |
||||||
|
Fujisawa et al, 2005[65] |
28 |
7 |
8 |
8 |
10 |
NR |
8 (29) |
|||||
|
Kameda et al, 2005[80] |
10 |
10 |
10 |
NR |
5 (50) |
|||||||
|
Takada et al, 2005[85] |
32 |
8 |
32 |
5 |
19 (59) |
10 (31) |
||||||
|
Wilkes et al, 2005[86] |
13 |
2 |
5 |
13 |
9 |
13 (100) |
0 (0) |
|||||
|
Ye et al, 2007[3] |
21 |
5 |
8 |
3 |
2 |
NR |
12 (57) |
|||||
|
Yamasaki et al, 2007[83] |
17 |
5 |
17 |
1 |
2 |
17 (17) |
0 (0) |
|||||
|
Ideura et al, 2007[64] |
18 |
3 |
2 |
13 |
3 |
13 (72) |
5 (28) |
|||||
|
Fathi et al, 2008[8] |
23 |
11 |
11 |
4 |
10 |
2 |
13/18 (72) |
2 (9) |
||||
|
Hayashi et al, 2008[5] |
33 |
25 |
8 |
NR |
9 (27) |
|||||||
|
Suzuki et al, 2009[89] |
5 |
4 |
5 |
5 |
2 (40) |
3 (60) |
||||||
|
Sem et al, 2009[87] |
11 |
8 |
7 |
4 |
2 |
11 |
1 |
7 (64) |
1 (9) |
|||
|
Kalluri et al, 2009[21] |
31 |
1 |
9 |
11 |
3 |
2 |
1 |
5 |
1 |
NR |
NR |
|
|
Mukae, 2009[94] |
27 |
8 |
8 |
4 |
1 |
2 |
NR |
6 (22) |
||||
|
Hervier et al, 2010[35] |
17 |
6 |
2 |
4 |
3 |
3 |
6/10 (60) |
0 (0) |
||||
|
Koreeda et al, 2010[14] |
14 |
6 |
1 |
1 |
6 |
12 (86) |
1 (8) |
|||||
|
Ji et al, 2010[7] |
69 |
23 |
16 |
7 |
6 |
2 |
36 |
NR |
24 (34) |
|||
|
Morganroth et al, 2010[82] |
4 |
4 |
4 (100) |
0 (0) |
||||||||
|
Lega et al, 2010[50] |
22 |
9 |
4 |
5 |
2 |
4 |
2 |
14 (64) |
1 (5) |
|||
|
Marie et al, 2011[67] |
107 |
56 |
25 |
38 |
6 |
90 (84) |
8 (7.5) |
|||||
|
Bakewell and Raghu, 2011[90] |
1 |
1 |
1 (100) |
1 (100) |
||||||||
|
Muro et al, 2012[46] |
10 |
5 |
3 |
6 |
10 (100) |
0 (0) |
||||||
|
Kunimasa et al, 2012[24] |
2 |
1 |
1 |
2 (100) |
2 (100) |
|||||||
|
Ingegnoli et al, 2012[84] |
15 |
3 |
8 |
4 |
4 (27) |
NR |
||||||
|
Cao et al, 2012[38] |
15 |
1 |
13 |
1 |
2 |
NR |
4 (27) |
|||||
|
Marie et al, 2013[15] |
66 |
31 |
17 |
27 |
6 |
55 (83) |
6 (9) |
Abbreviations: IVIG, intravenous immunoglobulin; NR, not reported.
Azathioprine has been used in patients with anti-Jo-1 ILD either as maintenance therapy following cyclophosphamide or as a steroid-sparing agent in patients receiving concomitant prednisone, with roughly 58% of patients demonstrating either resolution or significant improvement in pulmonary status.[15]
The safety of mycophenolate mofetil for the treatment of numerous connective tissue diseases is well established,[81] though its efficacy for ILD remains in question. There have been reports of DM-ILD patients with either significant improvement or complete resolution of their ILD following treatment with mycophenolate mofetil,[15] [82] including a case series of four patients who had improvement with mycophenolate mofetil after treatment with steroids and azathioprine proved to be ineffective.[67]
Cyclophosphamide, a nitrogen mustard alkylating agent, has also been used with some efficacy in treating myositis-related ILD.[15] [80] One study by Yamasaki et al used an intravenous dose of 300 to 800 mg/m2 at least six times every 4 weeks to treat refractory ILD and demonstrated both functional and radiographic improvement in the majority of patients.[83] Another study reported either resolution or a significant improvement in pulmonary status in 72% of anti-Jo-1 ILD patients treated with cyclophosphamide based on symptoms, PFTs, and HRCT imaging.[15] However, Ingegnoli et al observed improvement in only four of eight anti-Jo-1 PM ILD patients treated with a cyclophosphamide pulse plus steroids.[84]
Several studies have also highlighted a possible benefit of using cyclosporine A (CsA) to treat steroid-resistant ILD.[78] [85] Takada et al found that after 4 weeks of treatment, essentially all DM and PM patients diagnosed as having chronic ILD had at least some response to a combination of CsA and corticosteroids. Unfortunately, their results were less promising in those patients presenting with acute ILD; after 4 weeks of combination treatment, fewer than 50% of these patients had a partial response, and there were two reported deaths. Moreover, remission by the last clinical evaluation was only observed in roughly half the acute ILD patients, with an additional three deaths recorded over time. Interestingly, however, this study also seemed to suggest that patients who received a combination of CsA and steroids up front had better outcomes than those who initially received only steroids.[85] In contrast, Ingegnoli et al reported that five out of seven patients with anti-Jo-1-positive PM who were treated with CsA manifested CT evidence of worsening ILD, though the acuity of their ILD was not reported.[84]
There is evidence that the related calcineurin inhibitor tacrolimus may be superior to CsA, as several patients in the Takada study who had persistently active ILD despite prednisolone and CsA therapy demonstrated a prompt response to tacrolimus.[85] Other reports have also demonstrated the efficacy of tacrolimus in the treatment of anti-Jo-1-associated ILD.[86]
Rituximab, a monoclonal antibody directed against the B cell surface marker CD20, has demonstrated some efficacy in treating ILD in a group of anti-Jo-1-positive patients after 6 months in combination with prednisone; however, the true utility of rituximab was hard to evaluate since the majority of patients had received various other immunosuppressant agents both before and after rituximab was used.[87]
Based on evidence implicating a possible role for TNF-α in the immunopathology of IIM, investigators have also used the TNF inhibitor adalimumab to successfully treat Jo-1 antibody-positive ILD.[88] In addition, there have been reports of DM/PM-associated ILD that is refractory to high-dose steroids and CsA treated successfully with intravenous immunoglobulin,[89] [90] though significant data on the utility of such treatment are still lacking.
Clinical Course
Because myositis-associated ILD is a rare condition, most outcome analyses are reflective of case series or small, poorly controlled studies. Nevertheless, the available evidence suggests that outcomes in DM/PM and the antisynthetase syndrome are largely driven by the ILD phenotype, and to some extent, the presence of specific autoantibodies. Multiple reports have shown that patients with acute/subacute ILD in C-ADM often do not respond to therapy with corticosteroids and other immunosuppressive agents.[4] [10] [44] [91] By contrast, patients with radiologic or histologic evidence of an NSIP or COP pattern tend to be more responsive to steroid therapy.[15] [67] Furthermore, the presence of ARS antibodies in patients with ILD may predict greater steroid responsiveness, though with an increased risk of recurrence.[78]
Fathi et al followed patients with DM/PM ILD for 35 months after the initiation of treatment with corticosteroids plus another immunosuppressive agent and found that TLC improved in 38% of patients and deteriorated in 28%, with the remaining patients being labeled as stable. Interestingly, there were also several patients in whom PFTs normalized despite persistent radiographic abnormalities.[8] In a similar DM/PM population, Won Huh et al demonstrated increased FVC and DLCO 6 months following treatment, an improvement that was sustained for up to 3 years.[4] Likewise, Marie et al evaluated 107 patients with DM/PM ILD and found resolution in 32.7% and improvement in 51.4% with only 15.9% of patients deteriorating.[67] Koreeda et al reported improvement in 12 out of 14 patients with ARS antibodies treated with either steroids or CsA, though there was one case of progressive worsening and death despite an initial improvement at 6 months.[14]
Treatment results in ARS antibody-positive patients may be more favorable, with evidence that an NSIP or COP pattern on HRCT confers a greater probability of steroid responsiveness in anti-Jo-1 patients with ILD.[15] In a cohort of 28 anti-Jo-1 patients with ILD, for example, there was resolution in 25%, clinical improvement or stability in 54%, and worsening in 21%.[32] Other studies of anti-Jo-1 ILD patients have shown similar results, with remission noted in 29% of patients and improvement or stabilization in upward of two-thirds of patients, the majority of which were treated with either corticosteroids or a combination of corticosteroids plus immunosuppressive drugs.[26] [92] Furthermore, Marie et al demonstrated resolution of rapidly progressive, acute onset ILD in 8 of 12 anti-Jo-1 patients treated with steroids and either CsA, azathioprine, or mycophenolate.[15] Conversely, Ingegnoli et al showed less promising outcomes in a group of 15 patients with anti-Jo-1 PM; 60% had a worsening of their ILD 12 months after diagnosis.[84] Finally, although a group of patients with anti-PL-7 antibodies showed some improvement with treatment in 64.3% of cases, no patients experienced frank resolution—a pattern also seen in some patients with nonspecified DM/PM or anti-Jo-1-associated ILD.[34]
Overall, the biggest factor determining survival rates in patients with DM/PM and the antisynthetase syndrome is the presence of ILD. Marie et al demonstrated an overall mortality rate of 7.5% for myositis-associated ILD, with 87.5% of deaths attributable to the patients' lung disease.[67] Ji et al showed a DM/PM mortality rate of 34.8% in patients with ILD versus 8.6% in patients without ILD.[7] Similarly, the mortality rate among DM/PM patients in a Chinese cohort was 43.9% in patients with ILD versus 24.5% in patients without ILD.[6] Finally, Hayashi et al demonstrated a mortality rate of 12% in PM ILD patients versus 44% in DM ILD patients, with most of the deaths in the DM group attributable to rapidly progressive lung disease.[5] Indeed, acute ILD seems to portend a much worse prognosis than its chronic counterpart,[3] [7] with early (2 months) mortality rates as high as 72.7% in some cohorts of DM/PM ILD patients.[4]
Conclusions
The IIMs are a group of connective tissue diseases that include PM, DM, and ADM. Over the years, various MSAs have been identified, and their presence is associated with varying clinical phenotypes and propensities for the development of ILD. ILD in patients with DM/PM is associated with significant morbidity and mortality, and treatment typically consists of high-dose corticosteroids, often in conjunction with additional immunosuppressive agents. Although a majority of patients with ILD demonstrate either stability or improvement following treatment, there remains a subset of patients with fatal progression of their pulmonary disease. Further research in this field will require more detailed molecular profiling, more rigorous standardization of clinical data collection, improved consensus regarding outcome measures, and more balanced data sharing between institutions. The result will hopefully be a better understanding of the mechanisms underlying ILD, leading to more effective and targeted therapies.
-
References
- 1 Saketkoo LA, Ascherman DP, Cottin V, Christopher-Stine L, Danoff SK, Oddis CV. Interstitial lung disease in idiopathic inflammatory myopathy. Curr Rheumatol Rep 2010; 6 (2) 108-119
- 2 Chen IJ, Jan Wu YJ, Lin CW , et al. Interstitial lung disease in polymyositis and dermatomyositis. Clin Rheumatol 2009; 28 (6) 639-646
- 3 Ye S, Chen XX, Lu XY , et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol 2007; 26 (10) 1647-1654
- 4 Won Huh J, Soon Kim D, Keun Lee C , et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med 2007; 101 (8) 1761-1769
- 5 Hayashi S, Tanaka M, Kobayashi H , et al. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J Rheumatol 2008; 35 (2) 260-269
- 6 Yu KH, Wu YJ, Kuo CF , et al. Survival analysis of patients with dermatomyositis and polymyositis: analysis of 192 Chinese cases. Clin Rheumatol 2011; 30 (12) 1595-1601
- 7 Ji SY, Zeng FQ, Guo Q , et al. Predictive factors and unfavourable prognostic factors of interstitial lung disease in patients with polymyositis or dermatomyositis: a retrospective study. Chin Med J (Engl) 2010; 123 (5) 517-522
- 8 Fathi M, Vikgren J, Boijsen M , et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum 2008; 59 (5) 677-685
- 9 Chua F, Higton AM, Colebatch AN , et al. Idiopathic inflammatory myositis-associated interstitial lung disease: ethnicity differences and lung function trends in a British cohort. Rheumatology (Oxford) 2012; 51 (10) 1870-1876
- 10 Lee CS, Chen TL, Tzen CY , et al. Idiopathic inflammatory myopathy with diffuse alveolar damage. Clin Rheumatol 2002; 21 (5) 391-396
- 11 Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis 2004; 63 (3) 297-301
- 12 Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292 (8) 403-407
- 13 Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975; 292 (7) 344-347
- 14 Koreeda Y, Higashimoto I, Yamamoto M , et al. Clinical and pathological findings of interstitial lung disease patients with anti-aminoacyl-tRNA synthetase autoantibodies. Intern Med 2010; 49 (5) 361-369
- 15 Marie I, Josse S, Hatron PY , et al. Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res (Hoboken) 2013; 65 (5) 800-808
- 16 Sato S, Hirakata M, Kuwana M , et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52 (5) 1571-1576
- 17 Kaji K, Fujimoto M, Hasegawa M , et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46 (1) 25-28
- 18 Brouwer R, Hengstman GJ, Vree Egberts W , et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60 (2) 116-123
- 19 Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004; 50 (1) 209-215
- 20 Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with anti-synthetase autoantibodies. Intern Med J 2011; 41 (9) 674-679
- 21 Kalluri M, Sahn SA, Oddis CV , et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest 2009; 135 (6) 1550-1556
- 22 Hirakata M, Suwa A, Nagai S , et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol 1999; 162 (4) 2315-2320
- 23 Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007; 46 (6) 1005-1008
- 24 Kunimasa K, Arita M, Nakazawa T , et al. The clinical characteristics of two anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibody-positive interstitial lung disease patients with polymyositis/dermatomyositis. Intern Med 2012; 51 (24) 3405-3410
- 25 Hervier B, Meyer A, Dieval C , et al. Pulmonary hypertension in antisynthetase syndrome: prevalence, aetiology and survival. Eur Respir J 2013; 42 (5) 1271-1282
- 26 Marie I, Josse S, Decaux O , et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev 2012; 11 (10) 739-745
- 27 Hamaguchi Y, Fujimoto M, Matsushita T , et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS ONE 2013; 8 (4) e60442
- 28 Hall JC, Casciola-Rosen L, Samedy LA , et al. Anti-MDA5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken) 2013; 65 (8) 1307-1315
- 29 Hervier B, Benveniste O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr Rheumatol Rep 2013; 15 (8) 349
- 30 Gono T, Kawaguchi Y, Kuwana M , et al. Brief report: association of HLA-DRB1*0101/*0405 with susceptibility to anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis in the Japanese population. Arthritis Rheum 2012; 64 (11) 3736-3740
- 31 Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol 2007; 19 (6) 523-529
- 32 Marie I, Hatron PY, Dominique S , et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum 2012; 41 (6) 890-899
- 33 Richards TJ, Eggebeen A, Gibson K , et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum 2009; 60 (7) 2183-2192
- 34 Marie I, Josse S, Decaux O , et al. Clinical manifestations and outcome of anti-PL7 positive patients with antisynthetase syndrome. Eur J Intern Med 2013; 24 (5) 474-479
- 35 Hervier B, Wallaert B, Hachulla E , et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford) 2010; 49 (5) 972-976
- 36 Sato S, Hoshino K, Satoh T , et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60 (7) 2193-2200
- 37 Chen F, Wang D, Shu X, Nakashima R, Wang G. Anti-MDA5 antibody is associated with A/SIP and decreased T cells in peripheral blood and predicts poor prognosis of ILD in Chinese patients with dermatomyositis. Rheumatol Int 2012; 32 (12) 3909-3915
- 38 Cao H, Pan M, Kang Y , et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res (Hoboken) 2012; 64 (10) 1602-1610
- 39 Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011; 65 (1) 25-34
- 40 Kang EH, Nakashima R, Mimori T , et al. Myositis autoantibodies in Korean patients with inflammatory myositis: anti-140-kDa polypeptide antibody is primarily associated with rapidly progressive interstitial lung disease independent of clinically amyopathic dermatomyositis. BMC Musculoskelet Disord 2010; 11: 223
- 41 Koga T, Fujikawa K, Horai Y , et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford) 2012; 51 (7) 1278-1284
- 42 Fujikawa K, Kawakami A, Kaji K , et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: a single-centre, cross-sectional study. Scand J Rheumatol 2009; 38 (4) 263-267
- 43 Gono T, Sato S, Kawaguchi Y , et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology (Oxford) 2012; 51 (9) 1563-1570
- 44 Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology (Oxford) 2010; 49 (9) 1726-1733
- 45 Ikeda N, Takahashi K, Yamaguchi Y, Inasaka M, Kuwana M, Ikezawa Z. Analysis of dermatomyositis-specific autoantibodies and clinical characteristics in Japanese patients. J Dermatol 2011; 38 (10) 973-979
- 46 Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford) 2012; 51 (5) 800-804
- 47 Sato S, Kuwana M, Fujita T, Suzuki Y. Amyopathic dermatomyositis developing rapidly progressive interstitial lung disease with elevation of anti-CADM-140/MDA5 autoantibodies. Mod Rheumatol 2012; 22 (4) 625-629
- 48 Selva-O'Callaghan A, Labrador-Horrillo M, Solans-Laque R, Simeon-Aznar CP, Martínez-Gómez X, Vilardell-Tarrés M. Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients with idiopathic inflammatory myopathy. Arthritis Rheum 2006; 55 (5) 791-798
- 49 Venables PJ. Polymyositis-associated overlap syndromes. Br J Rheumatol 1996; 35 (4) 305-306
- 50 Lega JC, Cottin V, Fabien N, Thivolet-Béjui F, Cordier JF. Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-tRNA synthetase autoantibodies: a similar condition?. J Rheumatol 2010; 37 (5) 1000-1009
- 51 Ferreira JP, Almeida I, Marinho A, Cerveira C, Vasconcelos C. Anti-ro52 antibodies and interstitial lung disease in connective tissue diseases excluding scleroderma. ISRN Rheumatol 2012; 2012: 415272
- 52 Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109 (1) 32-40
- 53 Dugar M, Cox S, Limaye V, Gordon TP, Roberts-Thomson PJ. Diagnostic utility of anti-Ro52 detection in systemic autoimmunity. Postgrad Med J 2010; 86 (1012) 79-82
- 54 La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39 (3) 249-253
- 55 Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009; 48 (6) 607-612
- 56 Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum 1990; 33 (9) 1361-1370
- 57 Hengstman GJ, ter Laak HJ, Vree Egberts WT , et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis 2006; 65 (12) 1635-1638
- 58 Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73 (4) 420-428
- 59 Hengstman GJ, Vree Egberts WT, Seelig HP , et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis 2006; 65 (2) 242-245
- 60 Wang HB, Zhang Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res 2001; 29 (12) 2517-2521
- 61 Chinoy H, Salway F, Fertig N , et al; UK Adult Onset Myositis Immunogenetic Collaboration (AOMIC). In adult onset myositis, the presence of interstitial lung disease and myositis specific/associated antibodies are governed by HLA class II haplotype, rather than by myositis subtype. Arthritis Res Ther 2006; 8 (1) R13
- 62 Gono T, Kawaguchi Y, Satoh T , et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford) 2010; 49 (9) 1713-1719
- 63 Tanizawa K, Handa T, Nakashima R , et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir Med 2013; 107 (5) 745-752
- 64 Ideura G, Hanaoka M, Koizumi T , et al. Interstitial lung disease associated with amyopathic dermatomyositis: review of 18 cases. Respir Med 2007; 101 (7) 1406-1411
- 65 Fujisawa T, Suda T, Nakamura Y , et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol 2005; 32 (1) 58-64
- 66 Hervier B, Devilliers H, Stanciu R , et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 2012; 12 (2) 210-217
- 67 Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum 2011; 63 (11) 3439-3447
- 68 Fathi M, Barbasso Helmers S, Lundberg IE. KL-6: a serological biomarker for interstitial lung disease in patients with polymyositis and dermatomyositis. J Intern Med 2012; 271 (6) 589-597
- 69 Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years?. Chest 2010; 138 (6) 1464-1474
- 70 Le Goff B, Chérin P, Cantagrel A , et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum 2009; 61 (1) 108-118
- 71 Kono H, Inokuma S, Nakayama H, Suzuki M. Pneumomediastinum in dermatomyositis: association with cutaneous vasculopathy. Ann Rheum Dis 2000; 59 (5) 372-376
- 72 Korkmaz C, Ozkan R, Akay M, Hakan T. Pneumomediastinum and subcutaneous emphysema associated with dermatomyositis. Rheumatology (Oxford) 2001; 40 (4) 476-478
- 73 Maruoka H, Honda S, Takeo M , et al. A case of polymyositis complicated with interstitial pneumonitis and pneumomediastinum. Mod Rheumatol 2006; 16 (1) 55-57
- 74 Park SH, Kum YS, Kim KC, Choe JY, Park SH, Kim SK. Pneumomediastinum and subcutaneous emphysema secondary to amyopathic dermatomyositis with cryptogenic organizing pneumonia in invasive breast cancer: a case report and review of literature. Rheumatol Int 2009; 29 (10) 1231-1235
- 75 Powell C, Kendall B, Wernick R, Heffner JE. A 34-year-old man with amyopathic dermatomyositis and rapidly progressive dyspnea with facial swelling. Diagnosis: pneumomediastinum and subcutaneous emphysema secondary to amyopathic dermatomyositis-associated interstitial lung disease. Chest 2007; 132 (5) 1710-1713
- 76 Sandhya P, Keshava SN, Danda D, Padhan P, Mathew J, Gibikote S. Pneumorrhachis and pneumomediastinum in connective tissue disease-related interstitial lung disease: case series from a tertiary care teaching hospital in South India. Rheumatol Int 2012; 32 (5) 1415-1419
- 77 Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patients with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatology (Oxford) 2007; 46 (5) 842-845
- 78 Yoshifuji H, Fujii T, Kobayashi S , et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39 (3) 233-241
- 79 Hamaguchi Y, Kuwana M, Hoshino K , et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol 2011; 147 (4) 391-398
- 80 Kameda H, Nagasawa H, Ogawa H , et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol 2005; 32 (9) 1719-1726
- 81 Swigris JJ, Olson AL, Fischer A , et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest 2006; 130 (1) 30-36
- 82 Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken) 2010; 62 (10) 1496-1501
- 83 Yamasaki Y, Yamada H, Yamasaki M , et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46 (1) 124-130
- 84 Ingegnoli F, Lubatti C, Ingegnoli A, Boracchi P, Zeni S, Meroni PL. Interstitial lung disease outcomes by high-resolution computed tomography (HRCT) in Anti-Jo1 antibody-positive polymyositis patients: a single centre study and review of the literature. Autoimmun Rev 2012; 11 (5) 335-340
- 85 Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity 2005; 38 (5) 383-392
- 86 Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52 (8) 2439-2446
- 87 Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford) 2009; 48 (8) 968-971
- 88 Park JK, Yoo HG, Ahn DS, Jeon HS, Yoo WH. Successful treatment for conventional treatment-resistant dermatomyositis-associated interstitial lung disease with adalimumab. Rheumatol Int 2012; 32 (11) 3587-3590
- 89 Suzuki Y, Hayakawa H, Miwa S , et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung 2009; 187 (3) 201-206
- 90 Bakewell CJ, Raghu G. Polymyositis associated with severe interstitial lung disease: remission after three doses of IV immunoglobulin. Chest 2011; 139 (2) 441-443
- 91 Gono T, Kawaguchi Y, Hara M , et al. Increased ferritin predicts development and severity of acute interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford) 2010; 49 (7) 1354-1360
- 92 Tillie-Leblond I, Wislez M, Valeyre D , et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63 (1) 53-59
- 93 Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senécal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther 2007; 9 (4) R78
- 94 Mukae H, Ishimoto H, Sakamoto N , et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest 2009; 136: 1341-1347
Address for correspondence
-
References
- 1 Saketkoo LA, Ascherman DP, Cottin V, Christopher-Stine L, Danoff SK, Oddis CV. Interstitial lung disease in idiopathic inflammatory myopathy. Curr Rheumatol Rep 2010; 6 (2) 108-119
- 2 Chen IJ, Jan Wu YJ, Lin CW , et al. Interstitial lung disease in polymyositis and dermatomyositis. Clin Rheumatol 2009; 28 (6) 639-646
- 3 Ye S, Chen XX, Lu XY , et al. Adult clinically amyopathic dermatomyositis with rapid progressive interstitial lung disease: a retrospective cohort study. Clin Rheumatol 2007; 26 (10) 1647-1654
- 4 Won Huh J, Soon Kim D, Keun Lee C , et al. Two distinct clinical types of interstitial lung disease associated with polymyositis-dermatomyositis. Respir Med 2007; 101 (8) 1761-1769
- 5 Hayashi S, Tanaka M, Kobayashi H , et al. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J Rheumatol 2008; 35 (2) 260-269
- 6 Yu KH, Wu YJ, Kuo CF , et al. Survival analysis of patients with dermatomyositis and polymyositis: analysis of 192 Chinese cases. Clin Rheumatol 2011; 30 (12) 1595-1601
- 7 Ji SY, Zeng FQ, Guo Q , et al. Predictive factors and unfavourable prognostic factors of interstitial lung disease in patients with polymyositis or dermatomyositis: a retrospective study. Chin Med J (Engl) 2010; 123 (5) 517-522
- 8 Fathi M, Vikgren J, Boijsen M , et al. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum 2008; 59 (5) 677-685
- 9 Chua F, Higton AM, Colebatch AN , et al. Idiopathic inflammatory myositis-associated interstitial lung disease: ethnicity differences and lung function trends in a British cohort. Rheumatology (Oxford) 2012; 51 (10) 1870-1876
- 10 Lee CS, Chen TL, Tzen CY , et al. Idiopathic inflammatory myopathy with diffuse alveolar damage. Clin Rheumatol 2002; 21 (5) 391-396
- 11 Fathi M, Dastmalchi M, Rasmussen E, Lundberg IE, Tornling G. Interstitial lung disease, a common manifestation of newly diagnosed polymyositis and dermatomyositis. Ann Rheum Dis 2004; 63 (3) 297-301
- 12 Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292 (8) 403-407
- 13 Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975; 292 (7) 344-347
- 14 Koreeda Y, Higashimoto I, Yamamoto M , et al. Clinical and pathological findings of interstitial lung disease patients with anti-aminoacyl-tRNA synthetase autoantibodies. Intern Med 2010; 49 (5) 361-369
- 15 Marie I, Josse S, Hatron PY , et al. Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res (Hoboken) 2013; 65 (5) 800-808
- 16 Sato S, Hirakata M, Kuwana M , et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52 (5) 1571-1576
- 17 Kaji K, Fujimoto M, Hasegawa M , et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46 (1) 25-28
- 18 Brouwer R, Hengstman GJ, Vree Egberts W , et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001; 60 (2) 116-123
- 19 Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004; 50 (1) 209-215
- 20 Dugar M, Cox S, Limaye V, Blumbergs P, Roberts-Thomson PJ. Clinical heterogeneity and prognostic features of South Australian patients with anti-synthetase autoantibodies. Intern Med J 2011; 41 (9) 674-679
- 21 Kalluri M, Sahn SA, Oddis CV , et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest 2009; 135 (6) 1550-1556
- 22 Hirakata M, Suwa A, Nagai S , et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol 1999; 162 (4) 2315-2320
- 23 Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007; 46 (6) 1005-1008
- 24 Kunimasa K, Arita M, Nakazawa T , et al. The clinical characteristics of two anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibody-positive interstitial lung disease patients with polymyositis/dermatomyositis. Intern Med 2012; 51 (24) 3405-3410
- 25 Hervier B, Meyer A, Dieval C , et al. Pulmonary hypertension in antisynthetase syndrome: prevalence, aetiology and survival. Eur Respir J 2013; 42 (5) 1271-1282
- 26 Marie I, Josse S, Decaux O , et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev 2012; 11 (10) 739-745
- 27 Hamaguchi Y, Fujimoto M, Matsushita T , et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS ONE 2013; 8 (4) e60442
- 28 Hall JC, Casciola-Rosen L, Samedy LA , et al. Anti-MDA5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken) 2013; 65 (8) 1307-1315
- 29 Hervier B, Benveniste O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr Rheumatol Rep 2013; 15 (8) 349
- 30 Gono T, Kawaguchi Y, Kuwana M , et al. Brief report: association of HLA-DRB1*0101/*0405 with susceptibility to anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis in the Japanese population. Arthritis Rheum 2012; 64 (11) 3736-3740
- 31 Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol 2007; 19 (6) 523-529
- 32 Marie I, Hatron PY, Dominique S , et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum 2012; 41 (6) 890-899
- 33 Richards TJ, Eggebeen A, Gibson K , et al. Characterization and peripheral blood biomarker assessment of anti-Jo-1 antibody-positive interstitial lung disease. Arthritis Rheum 2009; 60 (7) 2183-2192
- 34 Marie I, Josse S, Decaux O , et al. Clinical manifestations and outcome of anti-PL7 positive patients with antisynthetase syndrome. Eur J Intern Med 2013; 24 (5) 474-479
- 35 Hervier B, Wallaert B, Hachulla E , et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatology (Oxford) 2010; 49 (5) 972-976
- 36 Sato S, Hoshino K, Satoh T , et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60 (7) 2193-2200
- 37 Chen F, Wang D, Shu X, Nakashima R, Wang G. Anti-MDA5 antibody is associated with A/SIP and decreased T cells in peripheral blood and predicts poor prognosis of ILD in Chinese patients with dermatomyositis. Rheumatol Int 2012; 32 (12) 3909-3915
- 38 Cao H, Pan M, Kang Y , et al. Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti-melanoma differentiation-associated gene 5 antibody. Arthritis Care Res (Hoboken) 2012; 64 (10) 1602-1610
- 39 Fiorentino D, Chung L, Zwerner J, Rosen A, Casciola-Rosen L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol 2011; 65 (1) 25-34
- 40 Kang EH, Nakashima R, Mimori T , et al. Myositis autoantibodies in Korean patients with inflammatory myositis: anti-140-kDa polypeptide antibody is primarily associated with rapidly progressive interstitial lung disease independent of clinically amyopathic dermatomyositis. BMC Musculoskelet Disord 2010; 11: 223
- 41 Koga T, Fujikawa K, Horai Y , et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford) 2012; 51 (7) 1278-1284
- 42 Fujikawa K, Kawakami A, Kaji K , et al. Association of distinct clinical subsets with myositis-specific autoantibodies towards anti-155/140-kDa polypeptides, anti-140-kDa polypeptides, and anti-aminoacyl tRNA synthetases in Japanese patients with dermatomyositis: a single-centre, cross-sectional study. Scand J Rheumatol 2009; 38 (4) 263-267
- 43 Gono T, Sato S, Kawaguchi Y , et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology (Oxford) 2012; 51 (9) 1563-1570
- 44 Hoshino K, Muro Y, Sugiura K, Tomita Y, Nakashima R, Mimori T. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology (Oxford) 2010; 49 (9) 1726-1733
- 45 Ikeda N, Takahashi K, Yamaguchi Y, Inasaka M, Kuwana M, Ikezawa Z. Analysis of dermatomyositis-specific autoantibodies and clinical characteristics in Japanese patients. J Dermatol 2011; 38 (10) 973-979
- 46 Muro Y, Sugiura K, Hoshino K, Akiyama M. Disappearance of anti-MDA-5 autoantibodies in clinically amyopathic DM/interstitial lung disease during disease remission. Rheumatology (Oxford) 2012; 51 (5) 800-804
- 47 Sato S, Kuwana M, Fujita T, Suzuki Y. Amyopathic dermatomyositis developing rapidly progressive interstitial lung disease with elevation of anti-CADM-140/MDA5 autoantibodies. Mod Rheumatol 2012; 22 (4) 625-629
- 48 Selva-O'Callaghan A, Labrador-Horrillo M, Solans-Laque R, Simeon-Aznar CP, Martínez-Gómez X, Vilardell-Tarrés M. Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients with idiopathic inflammatory myopathy. Arthritis Rheum 2006; 55 (5) 791-798
- 49 Venables PJ. Polymyositis-associated overlap syndromes. Br J Rheumatol 1996; 35 (4) 305-306
- 50 Lega JC, Cottin V, Fabien N, Thivolet-Béjui F, Cordier JF. Interstitial lung disease associated with anti-PM/Scl or anti-aminoacyl-tRNA synthetase autoantibodies: a similar condition?. J Rheumatol 2010; 37 (5) 1000-1009
- 51 Ferreira JP, Almeida I, Marinho A, Cerveira C, Vasconcelos C. Anti-ro52 antibodies and interstitial lung disease in connective tissue diseases excluding scleroderma. ISRN Rheumatol 2012; 2012: 415272
- 52 Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109 (1) 32-40
- 53 Dugar M, Cox S, Limaye V, Gordon TP, Roberts-Thomson PJ. Diagnostic utility of anti-Ro52 detection in systemic autoimmunity. Postgrad Med J 2010; 86 (1012) 79-82
- 54 La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006; 39 (3) 249-253
- 55 Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009; 48 (6) 607-612
- 56 Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum 1990; 33 (9) 1361-1370
- 57 Hengstman GJ, ter Laak HJ, Vree Egberts WT , et al. Anti-signal recognition particle autoantibodies: marker of a necrotising myopathy. Ann Rheum Dis 2006; 65 (12) 1635-1638
- 58 Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002; 73 (4) 420-428
- 59 Hengstman GJ, Vree Egberts WT, Seelig HP , et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis 2006; 65 (2) 242-245
- 60 Wang HB, Zhang Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res 2001; 29 (12) 2517-2521
- 61 Chinoy H, Salway F, Fertig N , et al; UK Adult Onset Myositis Immunogenetic Collaboration (AOMIC). In adult onset myositis, the presence of interstitial lung disease and myositis specific/associated antibodies are governed by HLA class II haplotype, rather than by myositis subtype. Arthritis Res Ther 2006; 8 (1) R13
- 62 Gono T, Kawaguchi Y, Satoh T , et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford) 2010; 49 (9) 1713-1719
- 63 Tanizawa K, Handa T, Nakashima R , et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir Med 2013; 107 (5) 745-752
- 64 Ideura G, Hanaoka M, Koizumi T , et al. Interstitial lung disease associated with amyopathic dermatomyositis: review of 18 cases. Respir Med 2007; 101 (7) 1406-1411
- 65 Fujisawa T, Suda T, Nakamura Y , et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis. J Rheumatol 2005; 32 (1) 58-64
- 66 Hervier B, Devilliers H, Stanciu R , et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 2012; 12 (2) 210-217
- 67 Marie I, Hatron PY, Dominique S, Cherin P, Mouthon L, Menard JF. Short-term and long-term outcomes of interstitial lung disease in polymyositis and dermatomyositis: a series of 107 patients. Arthritis Rheum 2011; 63 (11) 3439-3447
- 68 Fathi M, Barbasso Helmers S, Lundberg IE. KL-6: a serological biomarker for interstitial lung disease in patients with polymyositis and dermatomyositis. J Intern Med 2012; 271 (6) 589-597
- 69 Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years?. Chest 2010; 138 (6) 1464-1474
- 70 Le Goff B, Chérin P, Cantagrel A , et al. Pneumomediastinum in interstitial lung disease associated with dermatomyositis and polymyositis. Arthritis Rheum 2009; 61 (1) 108-118
- 71 Kono H, Inokuma S, Nakayama H, Suzuki M. Pneumomediastinum in dermatomyositis: association with cutaneous vasculopathy. Ann Rheum Dis 2000; 59 (5) 372-376
- 72 Korkmaz C, Ozkan R, Akay M, Hakan T. Pneumomediastinum and subcutaneous emphysema associated with dermatomyositis. Rheumatology (Oxford) 2001; 40 (4) 476-478
- 73 Maruoka H, Honda S, Takeo M , et al. A case of polymyositis complicated with interstitial pneumonitis and pneumomediastinum. Mod Rheumatol 2006; 16 (1) 55-57
- 74 Park SH, Kum YS, Kim KC, Choe JY, Park SH, Kim SK. Pneumomediastinum and subcutaneous emphysema secondary to amyopathic dermatomyositis with cryptogenic organizing pneumonia in invasive breast cancer: a case report and review of literature. Rheumatol Int 2009; 29 (10) 1231-1235
- 75 Powell C, Kendall B, Wernick R, Heffner JE. A 34-year-old man with amyopathic dermatomyositis and rapidly progressive dyspnea with facial swelling. Diagnosis: pneumomediastinum and subcutaneous emphysema secondary to amyopathic dermatomyositis-associated interstitial lung disease. Chest 2007; 132 (5) 1710-1713
- 76 Sandhya P, Keshava SN, Danda D, Padhan P, Mathew J, Gibikote S. Pneumorrhachis and pneumomediastinum in connective tissue disease-related interstitial lung disease: case series from a tertiary care teaching hospital in South India. Rheumatol Int 2012; 32 (5) 1415-1419
- 77 Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patients with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatology (Oxford) 2007; 46 (5) 842-845
- 78 Yoshifuji H, Fujii T, Kobayashi S , et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006; 39 (3) 233-241
- 79 Hamaguchi Y, Kuwana M, Hoshino K , et al. Clinical correlations with dermatomyositis-specific autoantibodies in adult Japanese patients with dermatomyositis: a multicenter cross-sectional study. Arch Dermatol 2011; 147 (4) 391-398
- 80 Kameda H, Nagasawa H, Ogawa H , et al. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J Rheumatol 2005; 32 (9) 1719-1726
- 81 Swigris JJ, Olson AL, Fischer A , et al. Mycophenolate mofetil is safe, well tolerated, and preserves lung function in patients with connective tissue disease-related interstitial lung disease. Chest 2006; 130 (1) 30-36
- 82 Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken) 2010; 62 (10) 1496-1501
- 83 Yamasaki Y, Yamada H, Yamasaki M , et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007; 46 (1) 124-130
- 84 Ingegnoli F, Lubatti C, Ingegnoli A, Boracchi P, Zeni S, Meroni PL. Interstitial lung disease outcomes by high-resolution computed tomography (HRCT) in Anti-Jo1 antibody-positive polymyositis patients: a single centre study and review of the literature. Autoimmun Rev 2012; 11 (5) 335-340
- 85 Takada K, Nagasaka K, Miyasaka N. Polymyositis/dermatomyositis and interstitial lung disease: a new therapeutic approach with T-cell-specific immunosuppressants. Autoimmunity 2005; 38 (5) 383-392
- 86 Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005; 52 (8) 2439-2446
- 87 Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford) 2009; 48 (8) 968-971
- 88 Park JK, Yoo HG, Ahn DS, Jeon HS, Yoo WH. Successful treatment for conventional treatment-resistant dermatomyositis-associated interstitial lung disease with adalimumab. Rheumatol Int 2012; 32 (11) 3587-3590
- 89 Suzuki Y, Hayakawa H, Miwa S , et al. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung 2009; 187 (3) 201-206
- 90 Bakewell CJ, Raghu G. Polymyositis associated with severe interstitial lung disease: remission after three doses of IV immunoglobulin. Chest 2011; 139 (2) 441-443
- 91 Gono T, Kawaguchi Y, Hara M , et al. Increased ferritin predicts development and severity of acute interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford) 2010; 49 (7) 1354-1360
- 92 Tillie-Leblond I, Wislez M, Valeyre D , et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008; 63 (1) 53-59
- 93 Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senécal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther 2007; 9 (4) R78
- 94 Mukae H, Ishimoto H, Sakamoto N , et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest 2009; 136: 1341-1347