
Subscribe to RSS
DOI: 10.1055/s-0030-1255560
© Georg Thieme Verlag KG Stuttgart · New York
Intravenöse Low-Dose-Immunglobulintherapie beim Skleromyxödem
Treatment of Scleromyxedema with Low-Dose Intravenous Immunoglobulin
Prof. Dr. med. Peter von den Driesch
Klinik für Dermatologie und Allergologie
Klinikum Stuttgart, Krankenhaus Bad Cannstatt
Prießnitzweg 24
70374 Stuttgart
Email: pdriesch@klinikum-stuttgart.de
Publication History
Publication Date:
29 July 2010 (online)

Zusammenfassung
Eine 53-jährige Patientin stellte sich mit lichenoid-papulösen, hämorrhagisch-bullösen und sklerodermieartigen Hautveränderungen an Armen und Beinen vor. Histologisch fand sich im Korium eine Fibrose und eine diffuse interstitielle Ansammlung von Muzinen. Es wurde die Diagnose eines Skleromyxödems gestellt. Eine monoklonale Gammopathie oder sichere systemische Manifestation lagen nicht vor. Unter Behandlung mit niedrig dosierten intravenösen Immunglobulinen (IVIG) kombiniert mit einem Kortikosteroidstoß war der klinische Befund regredient. Patienten mit Skleromyxödem können von einer niedrig dosierten IVIG-Therapie mit Kortikosteroidstoß profitieren. Größere Fallserien zur weiteren Evaluation sind erstrebenswert.
#Abstract
A 53-year-old woman presented with lichenoid-papular, hemorrhagic-bullous and scleroderma-like skin-lesions on her lower arms and lower legs. Skin biopsy showed dermal deposition of mucin and fibrosis. Scleromyxedema was diagnosed. No monoclonal gammopathy or definite systemic manifestations were observed. Intravenous immunoglobulin (IVIG) at a low dosage and systemic corticosteroids resulted in a remarkable response. IVIG at a low dosage in combination with systemic corticosteroids may be beneficial to patients with scleromyxedema. Larger series for further evaluation should be conducted.
#Einleitung
Das Skleromyxödem (Arndt-Gottron-Syndrom) ist eine seltene Erkrankung aus dem Formenkreis der Muzinosen, die charakterisiert ist durch einen sklerodermieartigen Aspekt, eine ausgeprägte Induration der Haut und lichenoide Papeln. Es findet sich fast regelmäßig eine Paraproteinämie. Häufig kommt es zu einer Manifestation der Erkrankung an inneren Organen [1]. Letale Verläufe wurden beschrieben [2].
Das Skleromyxödem kann als diffuse Form des Lichen myxödematosus aufgefasst werden. Daneben existiert dessen lokalisierte Form und eine dritte Variante, bei der u. a. ein Skleromyxödem ohne Paraproteinämie vorliegen kann [3].
Die histologischen Kriterien beinhalten typischerweise eine dermale Muzinansammlung sowie eine Fibroblastenproliferation und Anreicherung verdickter Kollagenbündel [4].
Die Behandlung des Skleromyxödems ist schwierig. Besonderes Interesse weckt der in den letzten Jahren zunehmend praktizierte Einsatz von IVIG.
#Kasuistik
Anamnese: Eine 53-jährige Patientin stellte sich mit einer seit drei Monaten bestehenden, progredienten „Hautverdickung” an den Extremitäten vor. Es bestand ein moderater Pruritus. Vorerkrankungen waren nicht bekannt.
Befund: Es fanden sich an Unterarmen und -schenkeln lokalisierte, teils lichenoid-papulöse, teils hämorrhagisch-bullöse Hautveränderungen bei einer sklerodermieartigen Induration der Haut ([Abb. 1] u. [2]).

Abb. 1 Sklerodermieartige Induration der Haut mit hämorrhagisch-bullösen Hautveränderungen und lichenoiden Papeln am rechten Unterarm bei Erstvorstellung.

Abb. 2 Sklerodermieartige Induration der Haut mit hämorrhagisch-bullösen Hautveränderungen und lichenoiden Papeln an den Unterschenkeln bei Erstvorstellung.
Labor: Neben dem Routinelabor wurden das ANA-Screening, die indirekte Immunfluoreszenz, die Immunfixationselektrophorese im Serum und Urinimmunfixationselektrophorese durchgeführt. Zudem wurden die Schilddrüsenparameter bestimmt und die Porphyriediagnostik sowie Borrelienserologie vorgenommen. Bis auf einen erhöhten Anti-Borrelien-IgG-Titer bestand kein sicher pathologischer Befund. Eine diskrete Proteinurie (12,9 mg/dl) ließ sich nach einigen Wochen nicht mehr nachweisen.
Neurologische Untersuchung: Es ergab sich kein Anhalt für eine Neuroborreliose.
Bildgebung: Im CT-Thorax/-Abdomen/-Becken fanden sich keine relevanten pathologischen Veränderungen. In den konventionellen Röntgenaufnahmen konnten keine Osteolysen detektiert werden. Echokardiografisch bestand eine leichtgradige Trikuspidalinsuffizienz.
Direkte Immunfluoreszenz: Die direkte Immunfluoreszenz war negativ.
Histologie: Zwei repräsentative Probeexzisionen zeigten eine subepitheliale Pseudoblasenbildung, im Korium eine massive Anreicherung von Fibroblasten und kollagenen Bindegewebsfasern sowie eine diffuse interstitielle Muzinansammlung ([Abb. 3] u. [4]).
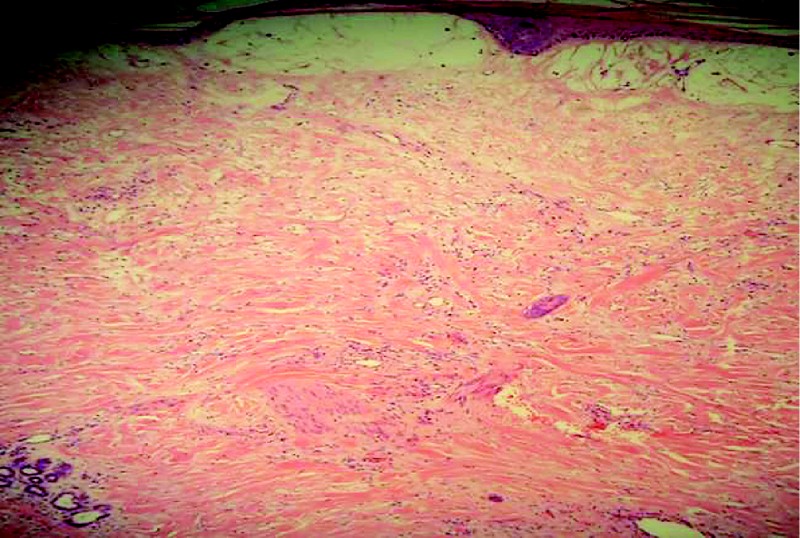
Abb. 3 Massive Anreicherung von Fibroblasten und kollagenen Bindegewebsfasern im Korium.

Abb. 4 Diffuse interstitielle Muzinansammlung im Korium.
Es wurde die Diagnose eines Skleromyxödems gestellt.
In einer ersten Sitzung wurde die IVIG-Gabe (Sandoglobulin® 0,1 g/kg KG pro Tag für fünf Tage) durchgeführt. In sechswöchigen Intervallen erfolgten fünf weitere je fünftägige IVIG-Applikationen in gleicher Dosierung. Kombiniert wurde jeweils ein Kortikosteroidstoß (Urbason® 60 – 80 mg p. o.) vorgenommen.
Unter der durchgeführten Therapie sistierte zunächst die Ausbildung neuer Hautveränderungen, im Verlauf heilten die Areale ab. Die sklerodermieartige Verdickung der Haut ließ inspektorisch ([Abb. 5] u. [6]) und palpatorisch nach. Es wurden keinerlei Nebenwirkungen beobachtet.

Abb. 5 Deutlich gebesserter Hautbefund am rechten Unterarm nach fünf Zyklen Low-Dose-IVIG.

Abb. 6 Deutlich gebesserter Hautbefund an den Unterschenkeln nach fünf Zyklen Low-Dose-IVIG.
Diskussion
Die Therapie des Skleromyxödems ist aufgrund dessen Rarität nur wenig evidenzbasiert und verläuft häufig frustran. In der Literatur wird über verschiedene Behandlungsmodalitäten berichtet. Meist handelt es sich dabei um Einzelfallbeschreibungen oder kleine Serien. Das Chemotherapeutikum Melphalan wurde eingesetzt. Der nachhaltige Therapieerfolg wird aber durch dessen Toxizität limitiert [1]. Nach extrakorporaler Photopherese wurden teils Rezidive beobachtet [5]. Glukokortikoide und weitere Immunsuppressiva wurden mit unterschiedlichem Erfolg angewendet. In jüngerer Zeit konnten nach Stammzelltransplantation vereinzelt längerfristig anhaltende Remissionen erzielt werden [6].
Es liegen mittlerweile aber auch mehrere ermutigende Veröffentlichungen über die Behandlung des Skleromyxödems mit IVIG vor. Insgesamt wird hier von hoher Effektivität bei minimalem Nebenwirkungsprofil berichtet [7] [8].
IVIG werden in der Dermatologie zur Therapie verschiedener autoimmunologischer Erkrankungen eingesetzt. Aufgrund der häufigen Assoziation mit einer Paraproteinämie ist eine autoimmunologische Genese auch beim Skleromyxödem wahrscheinlich. Die Applikation von IVIG zur Therapie des Skleromyxödems erscheint daher naheliegend.
In der Literatur finden sich unterschiedliche Angaben zur Dosierung von IVIG in der Therapie des Skleromyxödems. In einer Fallbeschreibung wurde eine Patientin mit einer Dosierung von 0,5 g/kg pro Tag für drei Tage mit zweimonatigem Therapieintervall bei einem Skleromyxödem ohne systemische Beteiligung behandelt [8]. In einer Serie wurden 2 g/kg als Gesamtdosis über 2 – 5 Tage verabreicht, wobei die Patienten hier häufig einen extrakutanen, systemischen Befall zeigten [9]. Wie der geschilderte Fall zeigt, profitieren Patienten bei der Immunglobulintherapie offenbar bereits von einer niedrigeren Gesamtdosis, insbesondere, wenn (noch) keine sicheren organischen Manifestationen vorliegen und kombiniert mit einem Kortikosteroid therapiert wird. Es bleibt abzuwarten, inwieweit diese Beobachtungen künftig auch bei der Behandlung anderer Patienten mit Skleromyxödem reproduzierbar sind. In einem solchen Falle würde sich die Frage stellen, ob durch ein noch weiteres Absenken der applizierten IVIG-Dosis ebenfalls noch ein therapeutischer Effekt erzielt werden könnte, und es wäre anzunehmen, dass die Dosierung intravenöser Immunglobuline in der Therapie auch anderer Autoimmunerkrankungen potenziell geringer gewählt werden könnte, als bisher angenommen.
Interessanterweise waren die Hautveränderungen in der Fallschilderung unter Immunglobulingabe reversibel, obgleich eine Paraproteinämie nicht nachgewiesen werden konnte. Dies ist ein Hinweis darauf, dass neben den Paraproteinen bzw. statt der Paraproteine andere Faktoren kausal für die Krankheitssymptome sind und durch die Immunglobulintherapie moduliert werden.
Es wird ein über das Fc-Fragment-mediierter Therapieeffekt der Immunglobuline angenommen; die genauen molekularbiologischen Mechanismen sind bislang ungeklärt. Im Falle des Skleromyxödems könnten Paraproteine und weitere Faktoren derart beeinflusst werden, dass es durch eine Inhibition von Makrophagen und Zytokinen zu verminderter Fibroblastenproliferation und Kollagensynthese sowie reduzierter Muzinproduktion kommt.
In der Literatur konnte ein Benefit für Patienten mit Skleromyxödem bei einer Kombination von Immunglobulinen mit einem systemischen Steroid beschrieben werden [10]. Dementsprechend wurde auch im geschilderten Fall additiv mit einem Steroidstoß therapiert. Unsere bisherigen Beobachtungen unterstützen die positiven Erfahrungen dieser anderen Autoren.
Es lässt sich festhalten, dass die IVIG-Gabe in der Therapie des Skleromyxödems offenbar eine effektive und durch ein minimales Nebenwirkungsprofil charakterisierte Behandlungsoption darstellt. Die Behandlung kann dabei als Low-dose-Applikation erfolgen. Auch das Skleromyxödem ohne Paraproteinämie scheint auf diese Therapieform anzusprechen. Eine Kombination mit systemischen Kortikosteroidstößen könnte sich in Kombination mit der IVIG-Gabe bewähren.
Größere Fallserien sind zur Evaluierung dieser Beobachtungen notwendig.
#Literatur
- 1 Dinneen A M, Dicken C H. Scleromyxedema. J Am Acad Dermatol. 1995; 33 37-43
- 2 Godby A, Bergstresser P R, Chaker B, Pandya A G. Fatal scleromyxedema: report of a case and review of the literature. J Am Acad Dermatol. 1998; 38 289-294
- 3 Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006; 25 100-104
- 4 Pomann J J, Rudner E J. Scleromyxedema revisited. Int J Dermatol. 2003; 42 31-35
- 5 Durani B K, Bock M, Naher H. Extracorporeal photopheresis – treatment option in scleromyxedema?. Hautarzt. 2001; 52 938-941
- 6 Lacy M Q, Hogan W J, Gertz M A. et al . Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation. Arch Dermatol. 2005; 141 1277-1282
- 7 Rey J B, Luria R B. Treatment of scleromyxedema and the dermatoneuro syndrome with intravenous immunoglobulin. J Am Acad Dermatol. 2009; 60 1037-1041
- 8 Lopez L, Wierzbicka-Hainaut E, Villers A, Guillet G. Efficacy of intravenous immunoglobulin in Arndt-Gottron scleromyxedema. Ann Dermatol Venereol. 2009; 136 330-336
- 9 Blum M, Wigley F M, Hummers L K. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine (Baltimore). 2008; 87 10-20
- 10 Majeski C, Taher M, Grewal P. et al . Combination oral prednisone and intravenous immunoglobulin in the treatment of scleromyxedema. J Cutan Med Surg. 2005; 9 99-104
Prof. Dr. med. Peter von den Driesch
Klinik für Dermatologie und Allergologie
Klinikum Stuttgart, Krankenhaus Bad Cannstatt
Prießnitzweg 24
70374 Stuttgart
Email: pdriesch@klinikum-stuttgart.de
Literatur
- 1 Dinneen A M, Dicken C H. Scleromyxedema. J Am Acad Dermatol. 1995; 33 37-43
- 2 Godby A, Bergstresser P R, Chaker B, Pandya A G. Fatal scleromyxedema: report of a case and review of the literature. J Am Acad Dermatol. 1998; 38 289-294
- 3 Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006; 25 100-104
- 4 Pomann J J, Rudner E J. Scleromyxedema revisited. Int J Dermatol. 2003; 42 31-35
- 5 Durani B K, Bock M, Naher H. Extracorporeal photopheresis – treatment option in scleromyxedema?. Hautarzt. 2001; 52 938-941
- 6 Lacy M Q, Hogan W J, Gertz M A. et al . Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation. Arch Dermatol. 2005; 141 1277-1282
- 7 Rey J B, Luria R B. Treatment of scleromyxedema and the dermatoneuro syndrome with intravenous immunoglobulin. J Am Acad Dermatol. 2009; 60 1037-1041
- 8 Lopez L, Wierzbicka-Hainaut E, Villers A, Guillet G. Efficacy of intravenous immunoglobulin in Arndt-Gottron scleromyxedema. Ann Dermatol Venereol. 2009; 136 330-336
- 9 Blum M, Wigley F M, Hummers L K. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine (Baltimore). 2008; 87 10-20
- 10 Majeski C, Taher M, Grewal P. et al . Combination oral prednisone and intravenous immunoglobulin in the treatment of scleromyxedema. J Cutan Med Surg. 2005; 9 99-104
Prof. Dr. med. Peter von den Driesch
Klinik für Dermatologie und Allergologie
Klinikum Stuttgart, Krankenhaus Bad Cannstatt
Prießnitzweg 24
70374 Stuttgart
Email: pdriesch@klinikum-stuttgart.de
Abb. 1 Sklerodermieartige Induration der Haut mit hämorrhagisch-bullösen Hautveränderungen und lichenoiden Papeln am rechten Unterarm bei Erstvorstellung.
Abb. 2 Sklerodermieartige Induration der Haut mit hämorrhagisch-bullösen Hautveränderungen und lichenoiden Papeln an den Unterschenkeln bei Erstvorstellung.
Abb. 3 Massive Anreicherung von Fibroblasten und kollagenen Bindegewebsfasern im Korium.
Abb. 4 Diffuse interstitielle Muzinansammlung im Korium.
Abb. 5 Deutlich gebesserter Hautbefund am rechten Unterarm nach fünf Zyklen Low-Dose-IVIG.
Abb. 6 Deutlich gebesserter Hautbefund an den Unterschenkeln nach fünf Zyklen Low-Dose-IVIG.