

Subscribe to RSS
DOI: 10.1055/a-2742-7859
Degradation of Cyclin-Dependent Kinase: A New Weapon for Cancer Therapy
Authors
Funding This study was financially supported by the Liaoning Innovative Talents in University (Grant No. LR2017043).


Abstract
Targeting cyclin-dependent kinase (CDK) families is a promising strategy for cancer therapy due to the close association between CDKs and an abnormal cell cycle or transcriptional regulation. However, after extensive clinical use, small molecule inhibitors of CDKs have also exposed issues, such as off-target effects or acquired drug resistance. Targeting protein degradation technology, which has been validated to be effective for many targets, has undergone more than 20 years of development, and some of these methods have been pushed into clinical trials. In this review, we summarized some successful reports on CDK-targeted degradation during recent years. Moreover, some challenging issues and future development trends are highlighted in the prospect section, which might provide updated insight into the development of novel CDK-targeted degraders with great potential as a new weapon for cancer therapy.
Introduction
The concept of the cell cycle and regulatory restriction points was first mentioned in the 1970s and early 1980s. In 2001, researchers behind the core work of identifying cyclin-dependent kinases (CDKs) and their partner cyclins in the cell cycle process were awarded the Nobel Prize.[1] [2] In recent years, with the continuous deepening of related research, dysregulation of cell growth and division has been proven to play a crucial role in the occurrence and development of cancers.[3] [4] [5] [6] Moreover, our understanding of how specific CDKs regulate transcription and maintain the oncogenic state has advanced considerably.[7] [8] [9] [10] Thus, CDK families have continuously become important targets for cancer treatment, which has led to considerable efforts to develop CDK inhibitors as cancer therapeutics. CDKs are Ser/Thr kinases that catalyze the phosphorylation of corresponding downstream proteins during the cell cycle and transcriptional regulation and usually combine with endogenous cyclin proteins to form a catalytic conformation. According to their different functions, CDKs can be divided into two main categories ([Fig. 1A]): cell cycle-associated CDKs (CDK1, CDK2, CDK4, and CDK6) and transcription-associated CDKs (CDK7, CDK8, CDK9, CDK12, CDK13, and CDK19).[11] [12] [13] [14]
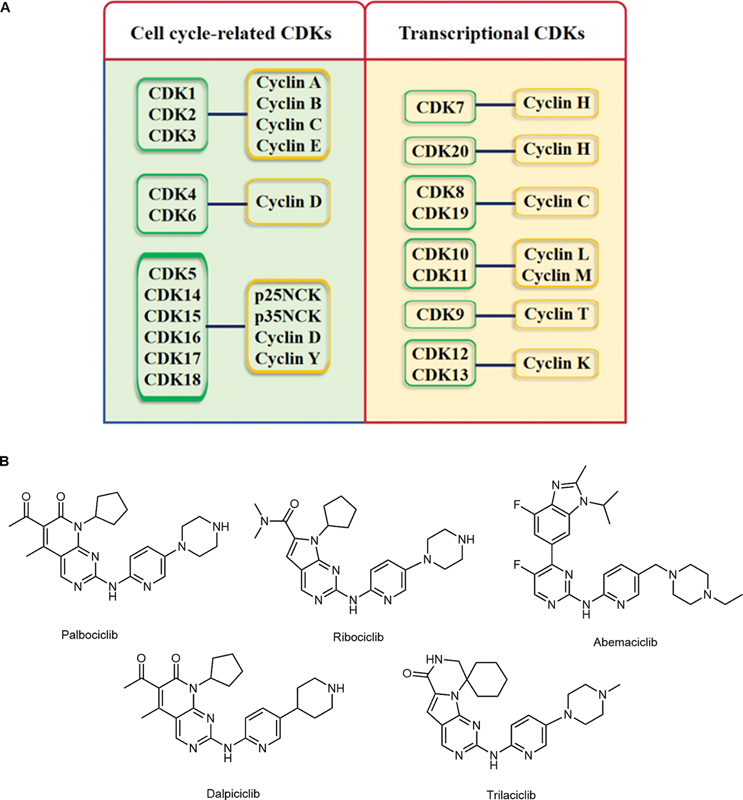
CDKs orchestrate oncogenic transformation through at least three interconnected mechanisms. First, mitogenic CDK4/6-cyclin D signaling initiates retinoblastoma (Rb) protein phosphorylation, liberating E2F factors to drive unscheduled G1–S transition and propagate chromosomal instability. Subsequent CDK2 and CDK1 activity enforces DNA re-replication and mitotic bypass, further amplifying genomic heterogeneity. Second, transcriptional CDK (notably CDK7, CDK9, and CDK12/13) govern RNA polymerase II pause release and productive elongation. This dependency sustains the expression of short-lived oncogenic transcripts (e.g., anti-apoptotic and growth factors), rendering tumors highly vulnerable to disruption of C-terminal domain (CTD) phosphorylation. Third, CDKs modulate processes such as DNA damage checkpoints, splicing, and translation, creating context-specific vulnerabilities. For example, CDK12 loss truncates homologous repair transcripts and induces PARP inhibitor sensitivity, whereas CDK9 inhibition preferentially depletes MCL-1 and intracisternal A-particle (IAP) mRNAs, promoting mitochondrial apoptosis. Together, these pleiotropic yet druggable functions establish CDKs as central executors of proliferative, transcriptional, and therapeutic liabilities in cancer.[15]
To date, the development of small molecule inhibitors of the CDK family has always been a popular research topic for scholars in the field of biological health. Starting from the first clinical CDK inhibitor, flavopiridol,[16] an increasing number of small molecule CDK inhibitors targeting different CDK subtypes are entering clinical research.[17] [18] [19] [20] [21] Among them, the CDK4/6 inhibitors palbociclib (from Pfizer), ribociclib (from Novartis), abemaciclib (from Lilly), dalpiciclib (from Jiangsu Hengrui), and trilaciclib (from G1 Therapeutics) were successively approved ([Fig. 1B]).[22] [23] [24] [25] [26] [27] However, with the widespread use of CDK inhibitors for clinical indications, some clinically observable issues and needs have also emerged, including side effects caused by poor CDK isoform selectivity, a lack of understanding of the precise mechanism of action, and hence, the absence of appropriate biomarkers and reduced tolerance caused by activation of other compensatory pathways.[28] [29] [30] [31] Briefly, the understanding and development of CDKs and the related drugs still require in-depth research.
The earliest research on targeting protein degradation (TPD) can be traced back to 2001. Sakamoto et al reported the peptide-based proteolysis targeting chimera (PROTAC) for the targeted degradation of methylthioamide peptidase 2 (METAP2), an enzyme known to effectively inhibit angiogenesis.[32] After more than two decades of development, significant breakthroughs have been made in TPD technology.[33] [34] [35] [36] Moreover, many TPD agents with different targets have successively entered clinical studies related to cancer treatment. The representative degradation agent Vepdegestrant (ARV-471), developed by Arvinas and Pfizer, entered phase III clinical trials.[37] [38] [39] [40] [41] The development and maturity of this technology also demonstrated breakthroughs and improvements in cancer therapeutic schedules. In this work, we reviewed recent research on the proteasome degradation of different CDK isoforms (2017–2025), which has focused mainly on PROTACs, molecular glues, and hydrophobic tag-based degraders. We aimed to analyze the feasibility of applying TPD technology to CDKs and to provide insights for future research.
Progress of PROTAC on Various Cyclin-Dependent Kinase Isoforms
PROTACs are heterobifunctional molecules composed of three modules: a target protein (protein of interest [POI]) ligand, an E3 ubiquitin ligase ligand, and a connecting linker. Their mechanism of action involves the simultaneous binding of the POI and an E3 ligase to form a POI–PROTAC–E3 ternary complex. This event recruits the E3 ligase to ubiquitinate the POI, marking it for recognition and degradation by the 26S proteasome. The PROTAC molecule is subsequently released and can catalyze multiple rounds of degradation. This strategy does not require occupancy of the target's active site, enabling the elimination of scaffold or structural proteins. Key advantages include their catalytic nature, potential to overcome drug resistance, and the ability to target undruggable proteins, positioning PROTACs as one of the most promising platforms for clinical translation in the field of targeted protein degradation.[35]
PROTACs of CDK2
CDK2, which is activated by the binding of cyclin E1, cyclin E2, and cyclin A2, is a core cell cycle regulator that is active in the G1 phase and throughout the S phase in dividing cells.[42] [43] [44] [45] Based on incomplete statistics, research describing the use of PROTAC degraders on CDK2 has been published. In 2021, Wang et al reported a first-in-class CDK2 selective degrader named CPS2 (1, [Fig. 2]) for acute myeloid leukemia (AML) differentiation therapy. In the construction of CPS2, J2 (a derivative of the CDK2 binder JNJ-7706621) and lenalidomide (a binder of CRBN) were chosen and linked by click chemistry.[46] CPS2 significantly inhibited CDK2 in AML cell lines (NB4, U937, and HL60) in a dose-dependent manner; however, the levels of CDK1, 4, 6, and 9 remained relatively unaffected, indicating good degradation selectivity of the degrader. Moreover, CPS2 markedly induced the differentiation of AML cell lines and primary patient cells, suggesting the potential of CPS2 for AML therapy and CDK2-related biomedical studies. In the same year, Hati et al identified PROTAC-8 (2, [Fig. 2B]), an AZD5438-based VHL-recruiting PROTAC that protects against cisplatin-induced ototoxicity and hearing loss, which created new opportunities for the application of PROTACs in cancer treatment and prognosis.[47] In 2025, Kwiatkowski's group reported compound 3 ([Fig. 3]),[48] an orally bioavailable and highly selective CDK2-targeting PROTAC discovered through structure-guided design. Compared with a clinical-stage CDK2 inhibitor, this degrader demonstrated superior phenotypic selectivity by preferentially eliminating CCNE1-amplified cancer cells while sparing nonamplified counterparts. In HCC169 xenograft models, a single dose sustained >90% CDK2 degradation and Rb phosphorylation inhibition, leading to durable tumor regression. In the same year, Kwiatkowski et al developed a highly selective CDK2 degrader, Cpd1 (4, [Fig. 3]), which unexpectedly also mediated the degradation of cyclin E1. Owing to its enhanced potency and selectivity for CDK2, this degrader drove antiproliferative activity with greater specificity against CCNE1-amplified cancer cells than small molecule inhibitors. Furthermore, the concurrent depletion of CDK2 and cyclin E1 resensitized palbociclib-adapted breast cancer cells to cell cycle arrest.[49]


PROTACs of CDK4/6
CDK4 and CDK6 (CDK4/6) are critical mediators of the cellular transition to S phase and are important for the initiation, growth, and survival of many cancer types. The best-documented function of CDK4/6-cyclin D in driving cell proliferation is the phosphorylation of the Rb protein, which leads to the release of E2Fs and the upregulation of E2F transcriptional targets.[50] [51] [52] [53] In 2019, the first PROTACs for CDK4/6 were obtained from palbociclib and ribociclib by Zhao and Burgess, which were named pal-pom (5, [Fig. 4]) and rib-pom (6, [Fig. 4]), respectively.[54] In the degradation ability test, palbociclib-based CRBN-recruiting PROTAC (5) showed better degradation of CDK4/6 than rib-pom (6) due to better kinase binding affinity, with DC50 values of 12.9 ± 3.5 nmol/L for CDK4 and 34.1 ± 7.3 nmol/L for CDK6 in the MDA-MB-231 cell line. Moreover, mechanistic research indicated that the CDK4/6-Rb-E2F axis was also downregulated due to the degradation of CDK4/6. Palbociclib-based PROTACs have been widely used for research on CDK4/6-related degradation or codegradation of other proteins. As shown in [Fig. 5], PROTAC 6 (7, [Fig. 5]),[55] BSJ-02-162 (8, [Fig. 5]),[56] CP-10 (9, [Fig. 5]),[57] and YX-2-107 (10, [Fig. 5])[58] were developed as palbociclib-based CRBN-recruiting PROTACs with various linkers by four different groups. Interestingly, PROTAC 6 (7, [Fig. 5]), CP-10 (9, [Fig. 5]), and YX-2-107 (10, [Fig. 5]) preferentially degraded CDK6 in the tested cell lines, with the DC50 values of 2.1 nmol/L (9 in the U251 cell line) and 4.4 nmol/L (10 in the BV173 cell line). Notably, in the research of CP-10 (9, [Fig. 5]), mutated and overexpressed CDK6 could still be degraded by CP-10, which might provide a new method for the treatment of mutant drug-resistant cancer. BSJ-02-162 (8, [Fig. 5]) was identified as a CDK4/6 dual-target degrader. Moreover, the results of quantitative proteomics showed that IKZF1/3 were also degraded synchronously. The authors believe that simultaneously degrading CDK4/6 and IKZF1/3 has an enhanced antiproliferative effect on MCL cell lines compared with that of palbociclib, lenalidomide, or a selective CDK4/6 degrader. Another thought-provoking aspect is that similar degradation agents might have varying abilities to degrade proteins in different cells, which is a future research direction. In 2025, He et al reported the rational design of a potent CDK4/6 protein degrader, compound 11 ([Fig. 5]). This compound achieved dual degradation of CDK4 and CDK6 proteins with DC50 values of 10.5 and 2.5 nmol/L, respectively. It also exhibited antiproliferative activity against Jurkat cells (IC50 = 0.18 μmol/L) and induced dose-dependent apoptosis and G1 phase cell cycle arrest.[59]


In addition to CRBN-recruiting PROTAC, several other E3 ubiquitin ligases and their ligands have also been reported for studying the degradation mechanism of CDK4/6. Anderson et al reported a palbociclib-based IAP-recruiting PROTAC (12, [Fig. 5]). Similar to compounds 7–9 mentioned above, 12 showed better degradation of CDK6 than of CDK4 in cells. However, this type of PROTAC might increase the sensitivity of cells that require IAPs for survival.[60] Moreover, Steinebach et al developed a novel selective CDK6 degrader (13, [Fig. 5]) based on palbociclib and a VHL ligand, which showed potent and long-lasting degradation in human and mouse cells and inhibited the proliferation of several leukemia, myeloma, and breast cancer cell lines.[61] Compared with that of 12, VHL-recruiting PROTAC 13 exhibited a better broad-spectrum degradation effect. In 2023, Pu et al reported a PROTAC degrader probe (14, [Fig. 5]) for CDK4/6 based on DCAF16 (DDB1- and CUL4-associated factor 16, mainly distributed in the nucleus), which showed potent inhibitory activity against CDK4/6 and decreased the level of the CDK4/6 protein in MDA-MB-231 cells in a concentration- and time-dependent manner. Moreover, the toxicity of 14 in normal cells was 7 times lower than that of palbociclib, and 14 exhibited therapeutic potential in MDA-MB-231 xenograft models in vivo.[62] This finding also indicates that the subcellular localization of E3 ubiquitin ligases may affect the degradation efficiency of PROTACs.
The codegradation of multiple proteins with a single compound provided the opportunity for collaborative cancer treatment. Verano et al reported the development of ALV-07-082-03 (15, [Fig. 5]), a CDK4/CDK6/Helios triple degrader that consists of palbociclib, which suppressed downstream CDK4/6 signaling and inhibited proliferation of cancer cells as well as enhanced immunomodulatory activity in comparison to the parental CDK4/6 inhibitor palbociclib or the selective Helios degrader DKY709.[63] Similarly, Xiong et al presented a novel PROTAC approach, termed bridged PROTAC, which utilizes a small-molecule binder of the target protein's binding partner to recruit the protein complex into proximity with an E3 ubiquitin ligase to target undruggable proteins. The novel PROTAC MS28 (16, [Fig. 5]) was successfully developed to effectively degrade cyclin D1 with faster degradation kinetics and superior degradation efficiency than CDK4/6 by recruiting the CDK4/6-cyclin D1 complex to the VHL E3 ligase, which provides a new strategy for the degradation of undruggable proteins that do not contain small-molecule binders.[64]
PROTACs of CDK7 and CDK8
CDK7 is a pivotal CDK family member with essential functions in cell cycle progression and transcription. Assembling with cyclin H and MAT1 in the cytoplasm to form the CAK complex, it activates CDKs 1, 2, 4, and 6 via T-loop phosphorylation to propel the cell cycle. Additionally, as part of the TFIIH complex, CDK7 directly contributes to transcriptional regulation.[65] [66] [67] [68] [69] CDK8, which might serve as a new cancer biomarker and is considered to be a potential target for cancer therapeutics, plays a vital role in regulating transcription either through its association with the mediator complex cyclin C or by phosphorylating transcription factors.[70] [71] [72] [73] [74] In 2024, Ji et al reported the first CDK7 degrader, JWZ-5-13 (17, [Fig. 6]), which was constructed by linking an ATP-competitive CDK7 ligand to a CRL2VHL E3 ligase recruiter. 17 effectively degraded CDK7 in a variety of cancer cell lines and significantly inhibited cell proliferation. Furthermore, it demonstrated favorable bioavailability in a mouse pharmacokinetic study.[75] Hatcher et al reported a novel CRBN-recruiting PROTAC, JH-XI-10-02 (18, [Fig. 6]), based on a derivative of cortistatin A, a natural product isolated from the marine sponge Corticium simplex that was identified as a potent and selective inhibitor of CDK8. Compound 18 significantly inhibited CDK8 in Jurkat cells. However, in CRBN-knockout Molt14 cells, CDK8 degradation was not observed. Although there are few relevant reports, PROTAC strategies targeting CDK8 still have significant development potential and research value.[76]

PROTACs of CDK9
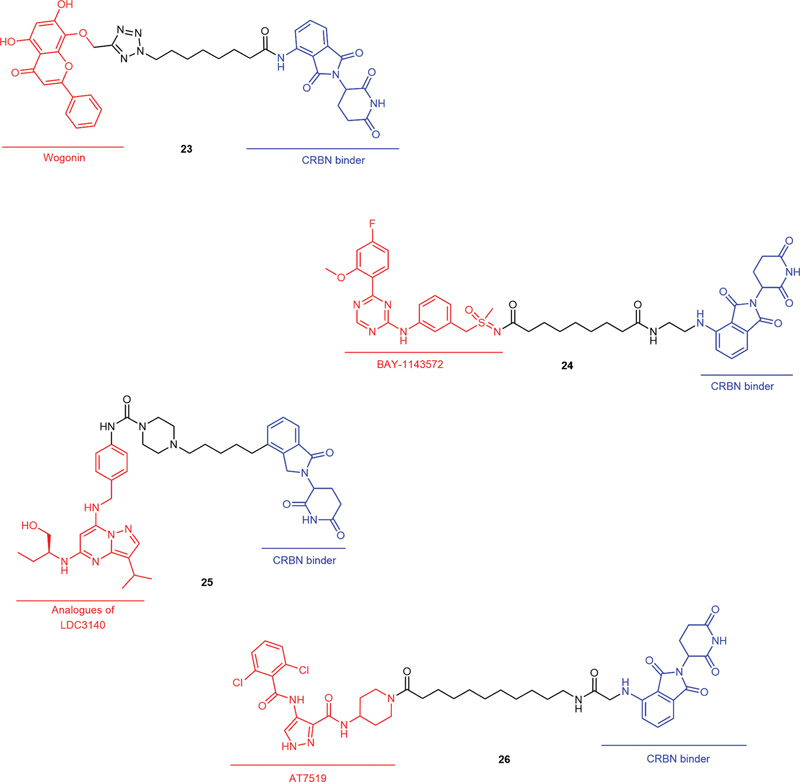
CDK9 is a member of the transcription CDK subfamily and plays a role in transcriptional regulation. CDK9 is mainly involved in the synthesis and processing of eukaryotic mRNA. Approximately 80% of CDK9 molecules form heterodimers with Cyclin T1, which is also called positive transcription elongation factor b (p-TEFb) and is required for the phosphorylation of the RNA polymerase II (RNA Pol II) CTD and transcription elongation. The remaining 20% of cells form complexes with Cyclin T2A, Cyclin T2B, or Cyclin K. CDK9 expression is dysregulated in various types of cancer. Inhibition or degradation of CDK9 results in transient transcriptional suppression and preferential depletion of apoptosis-related proteins with short half-lives, such as Mcl-1, c-Myc, and XIAP.[77] [78] [79] [80] Research on the development of CDK9 PROTACs seems to be quite popular, and the types of CDK9-binding ligands are also quite diverse. In 2017, Robb et al reported the first example of a PROTAC (19, [Fig. 7]) based on an aminopyrazole derivative that selectively degrades CDK9 while sparing other CDK family members in HCT116 cells, which resulted in reduced phosphorylation of Ser2 on RNA Pol II and decreased the levels of Mcl-1.[81] In another report, King et al discussed the influence of linker length on the degradation efficiency of an aminopyrazole-based CRBN-recruiting PROTAC. PROTAC 2 (20, [Fig. 7]) was found to selectively degrade CDK9 in MiaPaCa2 cells (DC50 = 158 ± 6 nmol/L) and to sensitize them to Venetoclax-mediated growth inhibition.[82] In 2018, THAL-SNS-032 (21, [Fig. 7]), a selective CRBN-recruiting CDK9 degrader developed from SNS-032, was reported to induce rapid degradation of CDK9 without affecting the levels of other SNS-032 targets. Moreover, other mechanistic studies have indicated that CDK9 degradation has prolonged cytotoxic effects compared with those of CDK9 inhibition.[83] Similarly, Pei et al reported SNS-032-based PROTAC bearing piperlongumine (PL) moiety (22, [Fig. 7]). In this work, PL was identified as a new covalent E3 ligase ligand of KEAP1, and PROTAC 22 was proven to induce CDK9 degradation in a ubiquitin–proteasome-dependent manner and to have more potent effects than SNS-032 against various tumor cells in vitro, these effects might be the driving force for the development of different types of CDK9 degraders.[84] In recent years, some other classic ligands of CDK9 were used in the construction of CRBN-recruiting PROTACs, including Wogonin (23, [Fig. 8]),[85] BAY-1143572 (24, [Fig. 8]),[86] analogues of LDC3140 (25, [Fig. 8])[87] and AT7519 (26, [Fig. 8]).[88] In 2024, Nie et al reported the design, synthesis, and extensive biological evaluation of a novel orally bioavailable, potent, and selective CDK9 degrader. The developed PROTAC demonstrated desirable potency (DC50 = 3.94 nmol/L) and high efficacy (Dmax = 96%) in degrading CDK9, effectively inhibiting the proliferation of triple-negative breast cancer (TNBC) MDA-MB-231 cells. PROTACs developed based on these ligands exhibited good and selective CDK9 degradation activity in the tested cells. Moreover, successful reports on the development of these CDK9-related PROTACs also provide new feasible solutions for the treatment of CDK9-related cancers.

PROTACs of CDK12/13
CDK12/cyclin K is a bifunctional complex implicated in cell division and transcription. Similar to CDK9, the active CDK12/cyclin K complex is recruited to the transcription start site by the polymerase II-associated factor 1 complex before elongation, which plays an indispensable role in the phosphorylation of the CTD of RNA Pol II during elongation.[89] [90] [91] In addition to transcription, CDK12 is also a key regulator of the transition from G1 to S phase, which is a vital step for DNA replication in the cell cycle.[92] [93] Thus, targeting CDK12 may become an ideal strategy for cancer treatment. However, existing CDK12 inhibitors potently inhibit the closest isoform of CDK13, which could cause potential toxicity.[94] [95] Therefore, the degradation of CDK12 or CDK12/13 may be more beneficial than inhibition.
In 2021, Jiang et al reported the first rational design and characterization of a CDK12-specific PROTAC named BSJ-4-116 (27, [Fig. 9]), which could selectively degrade CDK12 and result in premature cleavage and poly(adenylation) of DNA damage response (DDR) genes. Moreover, this work promoted the identification of two specific mutations in the G-loop of CDK12, Ile733Val and Gly739Ser, which might reduce the binding and efficacy of small molecule inhibitors or degradation agents.[96] Subsequently, Niu et al identified a potent CRBN-recruiting PROTAC degrader, PP-C8 (28, [Fig. 9]), based on the noncovalent dual inhibitor of CDK12/13, SR-4835, which showed high degradation selectivity toward CDK12, as well as its partner cyclin K. Importantly, 24-induced CDK12-cyclin K degradation suppresses DDR-related gene expression and shows a substantial synergistic antiproliferative effect with PARP inhibition in TNBC, which might overcome the current PARP inhibitor resistance in cancer treatment.[97] Additionally, in the field of TNBC treatment, Yang et al reported a highly potent and selective dual CDK12/13 PROTAC, compound 29 ([Fig. 10]), with DC50 values of 2.2 nmol/L (for CDK12) and 2.1 nmol/L (for CDK13) in MDA-MB-231 cells, respectively.[98] Moreover, 29 significantly improved the antiproliferative activity and suppressed the expression of DDR genes in a time- and dose-dependent manner in vitro. In 2024, the same research group designed and synthesized a novel CDK12/13 PROTAC degrader ZLC491 (30, [Fig. 10]) by introducing a rigid linker into the structure of 29. In TNBC MDA-MB-231 cells, this compound potently and concertedly degraded both CDK12 and CDK13, with DC50 values of 32 and 28 nmol/L, respectively. Global proteomic analysis and mechanistic studies confirmed that ZLC-491 selectively induces the degradation of CDK12/13 in a cereblom- and proteasome-dependent manner and exhibits an oral bioavailability of 46.8%.[99] That same year, researchers performed subsequent optimization based on 30, finding the orally bioavailable YJ1206 (31, [Fig. 10]) (IC50 = 12.55 nmol/L; oral bioavailability = 59.31%). This compound effectively suppressed the proliferation of prostate cancer cell subpopulations while sparing benign immortalized cells. Intriguingly, both degradation and genetic knockout of CDK12/13 led to activation of the AKT pathway. Combining CDK12/13 targeted degradation with AKT pathway inhibition resulted in a synthetic lethal effect in preclinical prostate cancer models.[100] Therefore, for targets with high homology but different therapeutic functions, synchronous protein degradation strategies may be more applicable than the design of selective small molecule inhibitors.


PROTACs of Multiple CDK Isoforms
Due to the high homology of some CDK isoforms, there are numerous difficulties associated with the development of a single inhibitor for a certain CDK subtype.[101] [102] Drawing on the successful experience of designing PROTACs for CDK12/13, the development of PROTACs with multiple CDK isoforms may overcome the shortcomings of a single CDK subtype of degradation agent, including off-target effects or acquired drug resistance.
In 2020, Teng et al developed the CDK2/5 dual degrader TMX-2172 (32, [Fig. 11]). In OVCAR8 cells (CCNE1 high expression), 32 showed significant degradation selectivity for CDK2 and CDK5 over other CDK isoforms, and the antiproliferative activity in OVCAR8 cells depended on CDK2 degradation. On the other hand, 32 may be a useful preliminary chemical tool for investigating CDK5-dependent biology in contexts where CDK2 is unlikely to play a role.[103] In the same year, Zhou et al identified a CDK2/9 dual degrader named F3 (33, [Fig. 11]), which potently induced the degradation of both CDK2 (DC50 = 62 nmol/L) and CDK9 (DC50 = 33 nmol/L) in human prostate cancer PC-3 cells.[104] In another study, Wei et al developed the first orally bioavailable prodrug of PROTAC based on ribociclib (34, [Fig. 11]) for degrading CDK2/4/6 in vivo.[105] The highlight of this work is the use of a prodrug strategy to improve the solubility and bioavailability of PROTACs in vivo while maintaining their strong degradation ability, which suggests that in the PROTAC design process, the drug properties of molecules should be fully considered.

Summary of PROTACs
The efficacy data of several representative PROTACs are summarized in [Table 1]. PROTAC degraders offer a unique mechanism of action, enabling the complete elimination of undruggable or drug-resistant proteins and overcoming mutation-driven escape. However, their development faces several challenges, including large molecular size, poor oral bioavailability, limited tissue selectivity, potential resistance risks, and insufficient long-term toxicity assessment. Moving forward, strategies such as artificial intelligence (AI)-assisted optimization of ternary complex kinetics to reduce molecular size, systematic screening of highly expressed E3 ligases to expand the ligand toolbox, and the development of prodrugs or antibody–PROTAC conjugates for improved delivery may help overcome these limitations.
Abbreviations: PROTAC, proteolysis targeting chimera; CDKs, cyclin-dependent kinases.
Advances in Molecular Glues Targeting CDK Isoforms
Molecular glues are a class of monofunctional small molecules that act by inducing novel, productive interactions between a target protein and an E3 ubiquitin ligase complex. Unlike bifunctional PROTACs, which physically link two proteins, molecular glues often function by binding to a shallow pocket on the E3 ligase or the target, thereby creating a new interfacial surface for recognition and binding. This event leads to the ubiquitination and proteasomal degradation of the target protein, effectively depleting it from the cell.[106] [107] Targeted protein degradation with molecular glue degraders has become a powerful therapeutic modality for eliminating classically undruggable disease-causing proteins through proteasome-mediated degradation.[108] [109] [110] However, the lack of universal molecular glue design principles and strategies limits the efficiency and applicability of molecular glue discovery. Even so, there are still some successful cases of CDK degradation through molecular glue strategies, which might provide some ideas for novel CDK-related molecular glue degraders.
Molecular Glue of CDK4
In 2023, Toriki et al identified a minimal covalent handle (but-2-ene-1,4-dione, fumarate derivative) using ribociclib as a prototype, which could work as a molecular glue (35, [Fig. 12]) to induce the proteasome-mediated degradation of CDK4 in cancer cells. Chemoproteomic profiling revealed interactions between molecular glue 35 and the covalent handle of RNF126 as well as other RING family E3 ligases, providing a novel strategy for converting protein-targeting ligands into covalent molecular glue degraders.[111]

Molecular Glue of CDK12/cyclin K
Compared with the therapeutic effects of PROTACs of CDK12, interference with the binding between CDK12 and its partner cyclin K or induction of cyclin K degradation seems to produce better therapeutic effects. Based on incomplete statistics, at least six articles demonstrated the effectiveness of the strategy. In 2020, Słabicki et al reported CR8 (36, [Fig. 13]), the first molecular glue degrader that depletes cyclin K.[112] The CDK12-binding form of 36 has a solvent-exposed pyridyl moiety that can induce the formation of a complex between CDK12–cyclin K and the CUL4 adaptor protein DDB1, which results in the ubiquitination and degradation of cyclin K and indirect inhibition of the function of CDK12. The binding mode of the DDB1–CDK12–cyclin K ternary complex with 36 is shown in [Fig. 13]. The phenylpyridine moiety of 36 is exposed to the solvent region of CDK12 and binds to the BPC domain of DDB1, which acts as a bridge between the CDK12/cyclin K complex and DDB1.

Based on the discovery of the CUL4–RBX1–DDB1 ligase core for the effective degradation of Cyclin K, several molecular glue degraders containing novel DDB1 binding pharmacophores for Cyclin K were developed, including HQ461 (37, [Fig. 14]),[113] NCT02 (38, [Fig. 14]),[114] 39 ([Fig. 14]),[115] SR-4835 (40, [Fig. 14]),[116] and derivatives of AT7519 (41, [Fig. 14]).[117] The successful development of these molecular glue degraders provides new insight for effectively regulating the function of CDKs, which indirectly disrupts the function of CDKs by degrading the related partner cyclins. Compared with PROTACs, molecular glue degraders possess advantages such as small molecular weights, good drug-like properties, low off-target effects, and low synthesis difficulty. However, how to rationally optimize molecular glue degraders is still unclear, and a better understanding of the molecular mechanisms and medicinal chemistry of molecular glue degraders is essential for translating targeted CDK degradation strategies into practical clinical applications.

Summary of Molecular Glue
Molecular glue degraders offer distinct advantages, including low molecular weight (typically <600 Da), high oral bioavailability, excellent cell membrane permeability, and straightforward synthesis. They function by inducing novel protein–protein interactions between an E3 ligase and a target protein, leading to ubiquitination and degradation, thereby holding potential for expanding the druggable target space. However, their discovery remains largely serendipitous, hindered by a limited repertoire of E3 ligands (primarily CRBN and DDB1), a lack of predictive models for ternary complex formation, and unclear mechanisms of target selectivity and resistance. To address these bottlenecks, future efforts should focus on establishing AI-assisted screening platforms integrated with high-throughput ubiquitination phenotyping, systematically expanding the E3 ligand toolbox, elucidating ternary complex structures, developing companion diagnostic strategies based on E3 genotypes, and exploring controllable prodrug approaches to enhance the systematic discovery and clinical translation of molecular glues.
Progress in the Development of Hydrophobic Tag-Based Degraders for Various CDK Isoforms
The use of a hydrophobic tag-based degrader (HyT-BD), which consists of a highly hydrophobic group and a small-molecule ligand of the target, is another promising strategy for targeted protein degradation. The hydrophobic tags exposed on the POI surface can be recognized as misfolded or damaged by endogenous chaperones, which results in degradation by the proteasome through protein quality control (PQC).[118] [119] [120] [121] [122] Compared with that of PROTACs and molecular glue degraders, the development of HyT-BD is in an early stage. Even so, some reports still demonstrate the effectiveness of applying hydrophobic tagging technology to induce CDK degradation.
Hydrophobic Tag-Based Degraders of CDK4/6
In 2022, Qiu et al reported the first hydrophobic tag-based degrader of CDK4/6 named LPM3770277 (42, [Fig. 15]), which consisted of abemaciclib and adamantane. Compound 42 induces CDK4/6 degradation via proteasome- and lysosome-promoted autophagy in a concentration- and time-dependent manner. Moreover, 42 showed superior antitumor efficacy and safety compared with those of abemaciclib in a TNBC xenograft model, which demonstrates the viability of using hydrophobic tag-based degraders as a strategy for developing potential treatments.[123]

Hydrophobic Tag-Based Degraders of CDK8 and CDK9
Wang's group reported the successive application of hydrophobic tag-based degradation strategies for CDK8/Cyclin C and CDK9/Cyclin T1. LL-K8-22 (43, [Fig. 15]) was identified as the first-in-class hydrophobic tag-based dual degrader of CDK8 and Cyclin C based on the structures of BI-1347 and Adamantane. Mechanistic research indicated that 43 could significantly degrade CDK8 and Cyclin C without reducing the levels of CDK19 or other cyclin proteins, which exhibited enhanced antiproliferative activity and more pronounced effects on CDK8–Cyclin C downstream signaling than BI-1347.[124] Furthermore, based on SNS-032, LL-K9-3[125] (44, [Fig. 15]) and LL-CDK9-12[126] (45, [Fig. 15]) were developed for the first application of hydrophobic tagging to induce the degradation of CDK9 and Cyclin T1 using menthol (44) and adamantane (45), respectively. Both degraders showed selective and synchronous degradation of CDK9 (DC50 = 0.662 μmol/L for 44 and DC50 = 0.362 μmol/L for 45) and Cyclin T1 (DC50 = 0.589 μmol/L for 44 and DC50 = 0.683 μmol/L for 45), which exerted stronger antiproliferative and proapoptotic effects than SNS-032 or THAL-SNS-032 (21), as mentioned above. In summary, HyT-BD represents a burgeoning protein degradation area that has the potential to be a new resource for small molecule drugs. However, this topic has not yet been fully explored. Discovering new hydrophobic tags with drug-likeness or auxiliary drug properties and novel applications of hydrophobic tags in CDKs or other cancer-related targets are still worthwhile research directions.
In 2025, our research group reported the first ATG101-recruiting CDK9 degrader, AZ-9 (46, [Fig. 15]), which was developed based on the hydrophobic tag-based Kinesin degradation technology. 46 showed significant degradation effects and selectivity toward other homologous cell cycle CDKs in vitro and in vivo, which could also affect downstream related phenotypes. Mechanism research revealed that AZ-9 recruits ATG101 to initiate the autophagy-lysosome pathway and forms autophagosomes through the recruitment of LC3, which then fuses with lysosomes to degrade CDK9 and the partner protein Cyclin T1.[127]
Summary of Hydrophobic Tag-Based Degraders
Hydrophobic tag (HyT)-based degraders function by fusing a target protein with a highly hydrophobic moiety, which mimics an unfolded state and leads to recognition by quality control systems (e.g., HSP70/CHIP) and subsequent proteasomal degradation. This approach offers advantages such as low molecular weight, straightforward synthesis, and independence from specific E3 ligases, making it applicable to challenging therapeutic targets. However, its efficacy is constrained by intrinsic protein folding stability and cellular quality control activity, often resulting in suboptimal selectivity, off-target degradation, and cytotoxicity. Additional limitations include rapid in vivo metabolism and a narrow therapeutic window. Current development is hampered by the lack of rational design principles, poor compatibility between the hydrophobic tag and protein binding sites, and insufficient control over in vivo delivery and release. Future efforts should integrate structural biology and computational modeling to optimize tag structure and attachment sites, develop controllable prodrug formats, screen for tissue-specific delivery systems, and establish evaluation standards focused on degradation selectivity to advance the preclinical translation of HyT degraders.
Discussion and Prospects
Since the first report of CDK degraders in 2017, research on CDK-related degradation agents has been ongoing for at least 8 years ([Fig. 16]), which has opened up another popular research field in addition to the development of small molecule inhibitors of CDKs. Numerous experiments have demonstrated the effectiveness of CDK degraders and their potential therapeutic advantages over small molecule inhibitors of CDKs. PROTACs, molecular glues, and hydrophobic tags (HyT) represent three distinct mechanistic paradigms for targeted protein degradation-“bifunctional recruitment,” “interface induction,” and “misfolding mimicry,” respectively. Each class offers unique advantages: the modular architecture of PROTACs enables precise target selectivity and access to “undruggable” proteins; molecular glues, typically under 600 Da, provide favorable oral bioavailability and synthetic tractability; and HyT degraders function independently of specific E3 ligases by co-opting the endogenous PQC machinery. However, their limitations are equally distinct. PROTACs are often hampered by high molecular weight, the hook effect, and a restricted E3 ligase repertoire. Molecular glue discovery remains largely serendipitous due to the absence of reliable predictive models for ternary complex formation. HyT degraders commonly suffer from poor selectivity, considerable cytotoxicity, and rapid in vivo clearance. The clinical translation of these technologies hinges on overcoming key bottlenecks: miniaturizing PROTACs and expanding the toolbox of tissue-specific E3 ligases; establishing AI- and structure-guided rational discovery platforms for molecular glues; and optimizing tag geometry and developing controlled delivery strategies for HyT degraders. Against this backdrop, the development of CDK degraders currently appears particularly promising. Their potential is such that they could, to a significant extent, surpass and eventually replace conventional small molecule CDK inhibitors.

Presently, from theory to practice, there are still some questions that need to be answered regarding CDK degraders as potential drugs for cancer treatment, which might be research directions worth considering in the future. The first noteworthy issue is the choice of CDK-binding ligands, which should possess high affinity and selectivity for related CDK isoforms. The selective development of small-molecule ligands or degradation agents for specific CDK isoforms is challenging due to the high homology of the CDK family. Targeting the inhibition or degradation of several different CDK isoforms, which might have synergistic effects and reverse the compensatory drug resistance caused by the influence of a single CDK subtype or produce unexpected side effects or toxicity, simultaneously has advantages and disadvantages for the treatment of cancer. Furthermore, selective effects on tumor tissue and normal tissue should be considered in the application of CDK degradation agents. The expression and function of related CDKs are different in normal and tumor tissues, even in different types of tumor tissues. Presently, the in vivo behavior of CDK degradation agents, including the pharmacokinetic properties of absorption, distribution, metabolism, and excretion, which may be affected by factors such as CDK ligand type, form of degradation agent, and linker, has not been fully elucidated. In recent years, the concept of a “Pro-degrader,” which can utilize the differential characteristics between normal and tumor tissues, such as different microenvironments or specific expressed proteins, for controlled drug release, has been proposed for precision medicine.[128] [129] [130] [131] [132] [133] This strategy could also be used in CDK degradation agents for enhanced selectivity.
The development of synthetic processes is another issue that needs to be considered for large-scale use of CDK degradation agents. Compared with those required by small molecule inhibitors, CDK degradation agents require more complex preparation processes due to their more complex molecular structures. However, there are few reports on the synthesis processes of degradation agents, and examples of the optimization of process parameters, traceability of related impurities, and establishment of analysis methods are still lacking.
CDK degraders have been validated to overcome the limitations of traditional inhibitors, including those related to catalytic site dependence and drug resistance, offering a novel therapeutic strategy for tumors driven by aberrant cell cycle progression or transcriptional addiction. We believe that the relevant technologies will continue to be optimized as research progresses, which could make CDK-related degradation agents effective weapons for cancer therapy instead of tools for chemical biology research.
Conflict of Interest
None declared.
-
References
- 1 Malumbres M. Cyclin-dependent kinases. Genome Biol 2014; 15 (06) 122
- 2 Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther 2017; 173: 83-105
- 3 Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol 2022; 19 (10) 619-636
- 4 Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M. Targeting DNA damage response pathways in cancer. Nat Rev Cancer 2023; 23 (02) 78-94
- 5 Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol 2022; 23 (01) 74-88
- 6 Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017; 17 (02) 93-115
- 7 Liu Y, Fu L, Wu J. et al. Transcriptional cyclin-dependent kinases: potential drug targets in cancer therapy. Eur J Med Chem 2022; 229: 114056
- 8 Parua PK, Fisher RP. Dissecting the Pol II transcription cycle and derailing cancer with CDK inhibitors. Nat Chem Biol 2020; 16 (07) 716-724
- 9 Simmons Kovacs LA, Orlando DA, Haase SB. Transcription networks and cyclin/CDKs: the yin and yang of cell cycle oscillators. Cell Cycle 2008; 7 (17) 2626-2629
- 10 Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 2005; 17 (09) 1033-1051
- 11 Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (02) 130-146
- 12 Roskoski Jr R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res 2019; 139: 471-488
- 13 Doonan JH, Kitsios G. Functional evolution of cyclin-dependent kinases. Mol Biotechnol 2009; 42 (01) 14-29
- 14 Xie Z, Hou S, Yang X. et al. Lessons learned from past cyclin-dependent kinase drug discovery efforts. J Med Chem 2022; 65 (09) 6356-6389
- 15 Pellarin I, Dall'Acqua A, Favero A. et al. Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct Target Ther 2025; 10 (01) 11
- 16 Ravishankar D, Rajora AK, Greco F, Osborn HM. Flavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol 2013; 45 (12) 2821-2831
- 17 Mounika P, Gurupadayya B, Kumar HY, Namitha B. An overview of CDK enzyme inhibitors in cancer therapy. Curr Cancer Drug Targets 2023; 23 (08) 603-619
- 18 Shi Z, Tian L, Qiang T. et al. From structure modification to drug launch: a systematic review of the ongoing development of cyclin-dependent kinase inhibitors for multiple cancer therapy. J Med Chem 2022; 65 (09) 6390-6418
- 19 Bhurta D, Bharate SB. Analyzing the scaffold diversity of cyclin-dependent kinase inhibitors and revisiting the clinical and preclinical pipeline. Med Res Rev 2022; 42 (02) 654-709
- 20 Jhaveri K, Burris Rd HA, Yap TA. et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev Anticancer Ther 2021; 21 (10) 1105-1124
- 21 Vidula N, Rugo HS. Cyclin-dependent kinase 4/6 inhibitors for the treatment of breast cancer: a review of preclinical and clinical data. Clin Breast Cancer 2016; 16 (01) 8-17
- 22 Hunter RJ, Park J, Asprer KJ, Doan AH. Updated review article: cyclin-dependent kinase 4/6 inhibitor impact, FDA approval, and resistance pathways. J Pharm Technol 2023; 39 (06) 298-308
- 23 Masuda N, Kosaka N, Iwata H, Toi M. Palbociclib as an early-line treatment for Japanese patients with hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: a review of clinical trial and real-world data. Int J Clin Oncol 2021; 26 (12) 2179-2193
- 24 Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clin Cancer Res 2017; 23 (13) 3251-3262
- 25 Kotake T, Toi M. Abemaciclib for the treatment of breast cancer. Expert Opin Pharmacother 2018; 19 (05) 517-524
- 26 Wang JR, Dong XT, Ashby CR. et al. Dalpiciclib. Cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, treatment of HR+/HER2-and HER2+advanced breast cancer. Drugs Future 2022; 47: 867-886
- 27 Qiu J, Sheng D, Lin F, Jiang P, Shi N. The efficacy and safety of trilaciclib in preventing chemotherapy-induced myelosuppression: a systematic review and meta-analysis of randomized controlled trials. Front Pharmacol 2023; 14: 1157251
- 28 Wild M, Hahn F, Brückner N. et al. Cyclin-dependent kinases (CDKs) and the human cytomegalovirus-encoded CDK ortholog pUL97 represent highly attractive targets for synergistic drug combinations. Int J Mol Sci 2022; 23 (05) 2493
- 29 Abdelmalak M, Singh R, Anwer M. et al. The renaissance of CDK inhibitors in breast cancer therapy: an update on clinical trials and therapy resistance. Cancers (Basel) 2022; 14 (21) 5388
- 30 Gomatou G, Trontzas I, Ioannou S, Drizou M, Syrigos N, Kotteas E. Mechanisms of resistance to cyclin-dependent kinase 4/6 inhibitors. Mol Biol Rep 2021; 48 (01) 915-925
- 31 Condorelli R, Spring L, O'Shaughnessy J. et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018; 29 (03) 640-645
- 32 Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. PROTACs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001; 98 (15) 8554-8559
- 33 Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022; 21 (03) 181-200
- 34 Li K, Crews CM. PROTACs: past, present and future. Chem Soc Rev 2022; 51 (12) 5214-5236
- 35 Cao C, He M, Wang L, He Y, Rao Y. Chemistries of bifunctional PROTAC degraders. Chem Soc Rev 2022; 51 (16) 7066-7114
- 36 Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B 2020; 10 (02) 207-238
- 37 Jiang H, Xiong H, Gu SX, Wang M. E3 ligase ligand optimization of clinical PROTACs. Front Chem 2023; 11: 1098331
- 38 Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol 2021; 12: 692574
- 39 Chirnomas D, Hornberger KR, Crews CM. Protein degraders enter the clinic - a new approach to cancer therapy. Nat Rev Clin Oncol 2023; 20 (04) 265-278
- 40 Campone M, Ma CX, Laurentiis MD. et al. VERITAC-2: a global, randomized phase 3 study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, vs fulvestrant in ER plus /human epidermal growth factor receptor 2 (HER2)-advanced breast cancer. J Clin Oncol 2023; 41: TPS1122
- 41 Layman RM, Jerzak KJ, Hilton JF. et al. TACTIVE-U: Phase 1b/2 umbrella study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, combined with other anticancer treatments in ER plus advanced or metastatic breast cancer. J Clin Oncol 2023; 41: TPS1121
- 42 Tadesse S, Caldon EC, Tilley W, Wang S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J Med Chem 2019; 62 (09) 4233-4251
- 43 Volkart PA, Bitencourt-Ferreira G, Souto AA, de Azevedo WF. Cyclin-dependent kinase 2 in cellular senescence and cancer. a structural and functional review. Curr Drug Targets 2019; 20 (07) 716-726
- 44 Chohan TA, Qian H, Pan Y, Chen JZ. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem 2015; 22 (02) 237-263
- 45 Woo RA, Poon RYC. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003; 2 (04) 316-324
- 46 Wang L, Shao X, Zhong T. et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat Chem Biol 2021; 17 (05) 567-575
- 47 Hati S, Zallocchi M, Hazlitt R. et al. AZD5438-PROTAC: a selective CDK2 degrader that protects against cisplatin- and noise-induced hearing loss. Eur J Med Chem 2021; 226: 113849
- 48 Collier PN, Zheng X, Ford M. et al. Discovery of selective and orally bioavailable heterobifunctional degraders of cyclin-dependent kinase 2. J Med Chem 2025; 68 (17) 18407-18422
- 49 Kwiatkowski N, Liang T, Sha Z. et al. CDK2 heterobifunctional degraders co-degrade CDK2 and cyclin E resulting in efficacy in CCNE1-amplified and overexpressed cancers. Cell Chem Biol 2025; 32 (04) 556-569.e24
- 50 Migliaccio I, Di Leo A, Malorni L. Cyclin-dependent kinase 4/6 inhibitors in breast cancer therapy. Curr Opin Oncol 2014; 26 (06) 568-575
- 51 Battisti NML, De Glas N, Sedrak MS. et al. Use of cyclin-dependent kinase 4/6 (CDK4/6) inhibitors in older patients with ER-positive HER2-negative breast cancer: Young International Society of Geriatric Oncology review paper. Ther Adv Med Oncol 2018; 10: 1758835918809610
- 52 Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 2017; 3 (01) 39-55
- 53 Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer 2022; 22 (06) 356-372
- 54 Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun (Camb) 2019; 55 (18) 2704-2707
- 55 Rana S, Bendjennat M, Kour S. et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett 2019; 29 (11) 1375-1379
- 56 Jiang B, Wang ES, Donovan KA. et al. Development of dual and selective degraders of Cyclin-dependent kinases 4 and 6. Angew Chem Int Ed Engl 2019; 58 (19) 6321-6326
- 57 Su S, Yang Z, Gao H. et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem 2019; 62 (16) 7575-7582
- 58 De Dominici M, Porazzi P, Xiao Y. et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020; 135 (18) 1560-1573
- 59 He H, Zhang X, Wang J. et al. Development of degraders of cyclin-dependent kinases 4 and 6 based on rational drug design. J Med Chem 2024; 67 (13) 11354-11364
- 60 Anderson NA, Cryan J, Ahmed A. et al. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg Med Chem Lett 2020; 30 (09) 127106
- 61 Steinebach C, Ng YLD, Sosič I. et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem Sci (Camb) 2020; 11 (13) 3474-3486
- 62 Pu C, Liu Y, Deng R. et al. Development of PROTAC degrader probe of CDK4/6 based on DCAF16. Bioorg Chem 2023; 138: 106637
- 63 Verano AL, You I, Donovan KA. et al. Redirecting the neo-substrate specificity of cereblon-targeting PROTACs to helios. ACS Chem Biol 2022; 17 (09) 2404-2410
- 64 Xiong Y, Zhong Y, Yim H. et al. Bridged proteolysis targeting chimera (PROTAC) enables degradation of undruggable targets. J Am Chem Soc 2022; 144 (49) 22622-22632
- 65 Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 2005; 118 (Pt 22): 5171-5180
- 66 Fisher RP. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019; 10 (02) 47-56
- 67 Larochelle S, Merrick KA, Terret ME. et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell 2007; 25 (06) 839-850
- 68 Schachter MM, Merrick KA, Larochelle S. et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell 2013; 50 (02) 250-260
- 69 Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011; 10 (07) 714-721
- 70 Menzl I, Witalisz-Siepracka A, Sexl V. CDK8-novel therapeutic opportunities. Pharmaceuticals (Basel) 2019; 12 (02) 92
- 71 Lv X, Tian Y, Li S. et al. Discovery and development of cyclin-dependent kinase 8 inhibitors. Curr Med Chem 2020; 27 (32) 5429-5443
- 72 Philip S, Kumarasiri M, Teo T, Yu M, Wang S. Cyclin-dependent kinase 8: a new hope in targeted cancer therapy? Miniperspective. J Med Chem 2018; 61 (12) 5073-5092
- 73 Fant CB, Taatjes DJ. Regulatory functions of the mediator kinases CDK8 and CDK19. Transcription 2019; 10 (02) 76-90
- 74 Xi M, Chen T, Wu C. et al. CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur J Med Chem 2019; 164: 77-91
- 75 Ji W, Du G, Jiang J. et al. Discovery of bivalent small molecule degraders of cyclin-dependent kinase 7 (CDK7). Eur J Med Chem 2024; 276: 116613
- 76 Hatcher JM, Wang ES, Johannessen L, Kwiatkowski N, Sim T, Gray NS. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett 2018; 9 (06) 540-545
- 77 Anshabo AT, Milne R, Wang S, Albrecht H. CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol 2021; 11: 678559
- 78 Shen YL, Wang YM, Zhang YX. et al. Targeting cyclin-dependent kinase 9 in cancer therapy. Acta Pharmacol Sin 2022; 43 (07) 1633-1645
- 79 Wu T, Qin Z, Tian Y. et al. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J Med Chem 2020; 63 (22) 13228-13257
- 80 Wu T, Wu X, Xu Y. et al. A patent review of selective CDK9 inhibitors in treating cancer. Expert Opin Ther Pat 2023; 33 (04) 309-322
- 81 Robb CM, Contreras JI, Kour S. et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 2017; 53 (54) 7577-7580
- 82 King HM, Rana S, Kubica SP. et al. Aminopyrazole based CDK9 PROTAC sensitizes pancreatic cancer cells to venetoclax. Bioorg Med Chem Lett 2021; 43: 128061
- 83 Olson CM, Jiang B, Erb MA. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol 2018; 14 (02) 163-170
- 84 Pei J, Xiao Y, Liu X. et al. Piperlongumine conjugates induce targeted protein degradation. Cell Chem Biol 2023; 30 (02) 203-213.e17
- 85 Bian J, Ren J, Li Y. et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem 2018; 81: 373-381
- 86 Qiu X, Li Y, Yu B. et al. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur J Med Chem 2021; 211: 113091
- 87 Wei D, Wang H, Zeng Q. et al. Discovery of potent and selective CDK9 degraders for targeting transcription regulation in triple-negative breast cancer. J Med Chem 2021; 64 (19) 14822-14847
- 88 Tokarski II RJ, Sharpe CM, Huntsman AC. et al. Bifunctional degraders of cyclin dependent kinase 9 (CDK9): probing the relationship between linker length, properties, and selective protein degradation. Eur J Med Chem 2023; 254: 115342
- 89 Chilà R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev 2016; 50: 83-88
- 90 Yan Z, Du Y, Zhang H. et al. Research progress of anticancer drugs targeting CDK12. RSC Med Chem 2023; 14 (09) 1629-1644
- 91 Liu H, Liu K, Dong Z. Targeting CDK12 for cancer therapy: function, mechanism, and drug discovery. Cancer Res 2021; 81 (01) 18-26
- 92 Choi SH, Kim S, Jones KA. Gene expression regulation by CDK12: a versatile kinase in cancer with functions beyond CTD phosphorylation. Exp Mol Med 2020; 52 (05) 762-771
- 93 Paculová H, Kohoutek J. The emerging roles of CDK12 in tumorigenesis. Cell Div 2017; 12: 7
- 94 Liang S, Hu L, Wu Z. et al. CDK12: a potent target and biomarker for human cancer therapy. Cells 2020; 9 (06) 1483
- 95 Tadesse S, Duckett DR, Monastyrskyi A. The promise and current status of CDK12/13 inhibition for the treatment of cancer. Future Med Chem 2021; 13 (02) 117-141
- 96 Jiang B, Gao Y, Che J. et al. Discovery and resistance mechanism of a selective CDK12 degrader. Nat Chem Biol 2021; 17 (06) 675-683
- 97 Niu T, Li K, Jiang L. et al. Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. Eur J Med Chem 2022; 228: 114012
- 98 Yang J, Chang Y, Tien JC. et al. Discovery of a highly potent and selective dual PROTAC degrader of CDK12 and CDK13. J Med Chem 2022; 65 (16) 11066-11083
- 99 Zhou L, Zhou K, Chang Y. et al. Discovery of ZLC491 as a potent, selective, and orally bioavailable CDK12/13 PROTAC degrader. J Med Chem 2024; 67 (20) 18247-18264
- 100 Chang Y, Wang X, Yang J. et al. Development of an orally bioavailable CDK12/13 degrader and induction of synthetic lethality with AKT pathway inhibition. Cell Rep Med 2024; 5 (10) 101752
- 101 Cheng W, Yang Z, Wang S. et al. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem 2019; 164: 615-639
- 102 Hardcastle IR, Golding BT, Griffin RJ. Designing inhibitors of cyclin-dependent kinases. Annu Rev Pharmacol Toxicol 2002; 42: 325-348
- 103 Teng M, Jiang J, He Z. et al. Development of CDK2 and CDK5 dual degrader TMX-2172. Angew Chem Int Ed Engl 2020; 59 (33) 13865-13870
- 104 Zhou F, Chen L, Cao C. et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem 2020; 187: 111952
- 105 Wei M, Zhao R, Cao Y. et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo . Eur J Med Chem 2021; 209: 112903
- 106 Dong G, Ding Y, He S, Sheng C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J Med Chem 2021; 64 (15) 10606-10620
- 107 Zhao L, Zhao J, Zhong K, Tong A, Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther 2022; 7 (01) 113
- 108 Wu H, Yao H, He C. et al. Molecular glues modulate protein functions by inducing protein aggregation: a promising therapeutic strategy of small molecules for disease treatment. Acta Pharm Sin B 2022; 12 (09) 3548-3566
- 109 Fang Y, He Q, Cao J. Targeted protein degradation and regulation with molecular glue: past and recent discoveries. Curr Med Chem 2022; 29 (14) 2490-2503
- 110 Domostegui A, Nieto-Barrado L, Perez-Lopez C, Mayor-Ruiz C. Chasing molecular glue degraders: screening approaches. Chem Soc Rev 2022; 51 (13) 5498-5517
- 111 Toriki ES, Papatzimas JW, Nishikawa K. et al. Rational chemical design of molecular glue degraders. ACS Cent Sci 2023; 9 (05) 915-926
- 112 Słabicki M, Kozicka Z, Petzold G. et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020; 585 (7824) 293-297
- 113 Lv L, Chen P, Cao L. et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 2020; 9: e59994
- 114 Dieter SM, Siegl C, Codó PL. et al. Degradation of CCNK/CDK12 is a druggable vulnerability of colorectal cancer. Cell Rep 2021; 36 (03) 109394
- 115 Jorda R, Havlíček L, Peřina M. et al. 3,5,7-substituted pyrazolo[4,3-d]pyrimidine inhibitors of cyclin-dependent kinases and Cyclin K degraders. J Med Chem 2022; 65 (13) 8881-8896
- 116 Houles T, Boucher J, Lavoie G. et al. The CDK12 inhibitor SR-4835 functions as a molecular glue that promotes cyclin K degradation in melanoma. Cell Death Discov 2023; 9 (01) 459
- 117 Thomas KL, Bouguenina H, Miller DSJ. et al. Degradation by design: new Cyclin K degraders from old CDK inhibitors. ACS Chem Biol 2024; 19 (01) 173-184
- 118 Peng X, Hu Z, Zeng L. et al. Overview of epigenetic degraders based on PROTAC, molecular glue, and hydrophobic tagging technologies. Acta Pharm Sin B 2024; 14 (02) 533-578
- 119 Ha S, Luo G, Xiang H. A comprehensive overview of small-molecule androgen receptor degraders: recent progress and future perspectives. J Med Chem 2022; 65 (24) 16128-16154
- 120 Kastl JM, Davies G, Godsman E, Holdgate GA. Small-molecule degraders beyond PROTACs challenges and opportunities. SLAS Discov 2021; 26 (04) 524-533
- 121 Xie S, Zhu J, Li J. et al. Small-molecule hydrophobic tagging: a promising strategy of druglike technology for targeted protein degradation. J Med Chem 2023; 66 (16) 10917-10933
- 122 He Q, Zhao X, Wu D. et al. Hydrophobic tag-based protein degradation: development, opportunity and challenge. Eur J Med Chem 2023; 260: 115741
- 123 Qiu J, Bai X, Zhang W. et al. LPM3770277, a potent novel CDK4/6 degrader, exerts antitumor effect against triple-negative breast cancer. Front Pharmacol 2022; 13: 853993
- 124 Wang M, Lin R, Li J. et al. Discovery of LL-K8–22: a selective, durable, and small-molecule degrader of the CDK8-cyclin C complex. J Med Chem 2023; 66 (07) 4932-4951
- 125 Li J, Liu T, Song Y. et al. Discovery of small-molecule degraders of the CDK9-Cyclin T1 complex for targeting transcriptional addiction in prostate cancer. J Med Chem 2022; 65 (16) 11034-11057
- 126 Lin R, Yang J, Liu T. et al. Discovery of HyT-based degraders of CDK9-Cyclin T1 complex. Chem Biodivers 2023; 20 (08) e202300769
- 127 Zhong Y, Xu J, Cao H. et al. First ATG101-recruiting small molecule degrader for selective CDK9 degradation via autophagy-lysosome pathway. Acta Pharm Sin B 2025; 15 (05) 2612-2624
- 128 Chen C, Yang Y, Wang Z, Li H, Dong C, Zhang X. Recent advances in Pro-PROTAC development to address on-target off-tumor toxicity. J Med Chem 2023; 66 (13) 8428-8440
- 129 Kounde CS, Shchepinova MM, Saunders CN. et al. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem Commun (Camb) 2020; 56 (41) 5532-5535
- 130 Cheng W, Li S, Wen X. et al. Development of hypoxia-activated PROTAC exerting a more potent effect in tumor hypoxia than in normoxia. Chem Commun (Camb) 2021; 57 (95) 12852-12855
- 131 Shi S, Du Y, Zou Y. et al. Rational design for nitroreductase (NTR)-responsive proteolysis targeting chimeras (PROTACs) selectively targeting tumor tissues. J Med Chem 2022; 65 (06) 5057-5071
- 132 Reynders M, Matsuura BS, Bérouti M. et al. PHOTACs enable optical control of protein degradation. Sci Adv 2020; 6 (08) eaay5064
- 133 Pfaff P, Samarasinghe KTG, Crews CM, Carreira EM. Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent Sci 2019; 5 (10) 1682-1690
Address for correspondence
Publication History
Received: 14 April 2025
Accepted: 10 November 2025
Article published online:
11 December 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Malumbres M. Cyclin-dependent kinases. Genome Biol 2014; 15 (06) 122
- 2 Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther 2017; 173: 83-105
- 3 Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol 2022; 19 (10) 619-636
- 4 Groelly FJ, Fawkes M, Dagg RA, Blackford AN, Tarsounas M. Targeting DNA damage response pathways in cancer. Nat Rev Cancer 2023; 23 (02) 78-94
- 5 Matthews HK, Bertoli C, de Bruin RAM. Cell cycle control in cancer. Nat Rev Mol Cell Biol 2022; 23 (01) 74-88
- 6 Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017; 17 (02) 93-115
- 7 Liu Y, Fu L, Wu J. et al. Transcriptional cyclin-dependent kinases: potential drug targets in cancer therapy. Eur J Med Chem 2022; 229: 114056
- 8 Parua PK, Fisher RP. Dissecting the Pol II transcription cycle and derailing cancer with CDK inhibitors. Nat Chem Biol 2020; 16 (07) 716-724
- 9 Simmons Kovacs LA, Orlando DA, Haase SB. Transcription networks and cyclin/CDKs: the yin and yang of cell cycle oscillators. Cell Cycle 2008; 7 (17) 2626-2629
- 10 Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 2005; 17 (09) 1033-1051
- 11 Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (02) 130-146
- 12 Roskoski Jr R. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol Res 2019; 139: 471-488
- 13 Doonan JH, Kitsios G. Functional evolution of cyclin-dependent kinases. Mol Biotechnol 2009; 42 (01) 14-29
- 14 Xie Z, Hou S, Yang X. et al. Lessons learned from past cyclin-dependent kinase drug discovery efforts. J Med Chem 2022; 65 (09) 6356-6389
- 15 Pellarin I, Dall'Acqua A, Favero A. et al. Cyclin-dependent protein kinases and cell cycle regulation in biology and disease. Signal Transduct Target Ther 2025; 10 (01) 11
- 16 Ravishankar D, Rajora AK, Greco F, Osborn HM. Flavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol 2013; 45 (12) 2821-2831
- 17 Mounika P, Gurupadayya B, Kumar HY, Namitha B. An overview of CDK enzyme inhibitors in cancer therapy. Curr Cancer Drug Targets 2023; 23 (08) 603-619
- 18 Shi Z, Tian L, Qiang T. et al. From structure modification to drug launch: a systematic review of the ongoing development of cyclin-dependent kinase inhibitors for multiple cancer therapy. J Med Chem 2022; 65 (09) 6390-6418
- 19 Bhurta D, Bharate SB. Analyzing the scaffold diversity of cyclin-dependent kinase inhibitors and revisiting the clinical and preclinical pipeline. Med Res Rev 2022; 42 (02) 654-709
- 20 Jhaveri K, Burris Rd HA, Yap TA. et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev Anticancer Ther 2021; 21 (10) 1105-1124
- 21 Vidula N, Rugo HS. Cyclin-dependent kinase 4/6 inhibitors for the treatment of breast cancer: a review of preclinical and clinical data. Clin Breast Cancer 2016; 16 (01) 8-17
- 22 Hunter RJ, Park J, Asprer KJ, Doan AH. Updated review article: cyclin-dependent kinase 4/6 inhibitor impact, FDA approval, and resistance pathways. J Pharm Technol 2023; 39 (06) 298-308
- 23 Masuda N, Kosaka N, Iwata H, Toi M. Palbociclib as an early-line treatment for Japanese patients with hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: a review of clinical trial and real-world data. Int J Clin Oncol 2021; 26 (12) 2179-2193
- 24 Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clin Cancer Res 2017; 23 (13) 3251-3262
- 25 Kotake T, Toi M. Abemaciclib for the treatment of breast cancer. Expert Opin Pharmacother 2018; 19 (05) 517-524
- 26 Wang JR, Dong XT, Ashby CR. et al. Dalpiciclib. Cyclin-dependent kinase 4/6 (CDK4/6) inhibitor, treatment of HR+/HER2-and HER2+advanced breast cancer. Drugs Future 2022; 47: 867-886
- 27 Qiu J, Sheng D, Lin F, Jiang P, Shi N. The efficacy and safety of trilaciclib in preventing chemotherapy-induced myelosuppression: a systematic review and meta-analysis of randomized controlled trials. Front Pharmacol 2023; 14: 1157251
- 28 Wild M, Hahn F, Brückner N. et al. Cyclin-dependent kinases (CDKs) and the human cytomegalovirus-encoded CDK ortholog pUL97 represent highly attractive targets for synergistic drug combinations. Int J Mol Sci 2022; 23 (05) 2493
- 29 Abdelmalak M, Singh R, Anwer M. et al. The renaissance of CDK inhibitors in breast cancer therapy: an update on clinical trials and therapy resistance. Cancers (Basel) 2022; 14 (21) 5388
- 30 Gomatou G, Trontzas I, Ioannou S, Drizou M, Syrigos N, Kotteas E. Mechanisms of resistance to cyclin-dependent kinase 4/6 inhibitors. Mol Biol Rep 2021; 48 (01) 915-925
- 31 Condorelli R, Spring L, O'Shaughnessy J. et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018; 29 (03) 640-645
- 32 Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. PROTACs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001; 98 (15) 8554-8559
- 33 Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022; 21 (03) 181-200
- 34 Li K, Crews CM. PROTACs: past, present and future. Chem Soc Rev 2022; 51 (12) 5214-5236
- 35 Cao C, He M, Wang L, He Y, Rao Y. Chemistries of bifunctional PROTAC degraders. Chem Soc Rev 2022; 51 (16) 7066-7114
- 36 Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B 2020; 10 (02) 207-238
- 37 Jiang H, Xiong H, Gu SX, Wang M. E3 ligase ligand optimization of clinical PROTACs. Front Chem 2023; 11: 1098331
- 38 Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol 2021; 12: 692574
- 39 Chirnomas D, Hornberger KR, Crews CM. Protein degraders enter the clinic - a new approach to cancer therapy. Nat Rev Clin Oncol 2023; 20 (04) 265-278
- 40 Campone M, Ma CX, Laurentiis MD. et al. VERITAC-2: a global, randomized phase 3 study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, vs fulvestrant in ER plus /human epidermal growth factor receptor 2 (HER2)-advanced breast cancer. J Clin Oncol 2023; 41: TPS1122
- 41 Layman RM, Jerzak KJ, Hilton JF. et al. TACTIVE-U: Phase 1b/2 umbrella study of ARV-471, a proteolysis targeting chimera (PROTAC) estrogen receptor (ER) degrader, combined with other anticancer treatments in ER plus advanced or metastatic breast cancer. J Clin Oncol 2023; 41: TPS1121
- 42 Tadesse S, Caldon EC, Tilley W, Wang S. Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J Med Chem 2019; 62 (09) 4233-4251
- 43 Volkart PA, Bitencourt-Ferreira G, Souto AA, de Azevedo WF. Cyclin-dependent kinase 2 in cellular senescence and cancer. a structural and functional review. Curr Drug Targets 2019; 20 (07) 716-726
- 44 Chohan TA, Qian H, Pan Y, Chen JZ. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem 2015; 22 (02) 237-263
- 45 Woo RA, Poon RYC. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003; 2 (04) 316-324
- 46 Wang L, Shao X, Zhong T. et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat Chem Biol 2021; 17 (05) 567-575
- 47 Hati S, Zallocchi M, Hazlitt R. et al. AZD5438-PROTAC: a selective CDK2 degrader that protects against cisplatin- and noise-induced hearing loss. Eur J Med Chem 2021; 226: 113849
- 48 Collier PN, Zheng X, Ford M. et al. Discovery of selective and orally bioavailable heterobifunctional degraders of cyclin-dependent kinase 2. J Med Chem 2025; 68 (17) 18407-18422
- 49 Kwiatkowski N, Liang T, Sha Z. et al. CDK2 heterobifunctional degraders co-degrade CDK2 and cyclin E resulting in efficacy in CCNE1-amplified and overexpressed cancers. Cell Chem Biol 2025; 32 (04) 556-569.e24
- 50 Migliaccio I, Di Leo A, Malorni L. Cyclin-dependent kinase 4/6 inhibitors in breast cancer therapy. Curr Opin Oncol 2014; 26 (06) 568-575
- 51 Battisti NML, De Glas N, Sedrak MS. et al. Use of cyclin-dependent kinase 4/6 (CDK4/6) inhibitors in older patients with ER-positive HER2-negative breast cancer: Young International Society of Geriatric Oncology review paper. Ther Adv Med Oncol 2018; 10: 1758835918809610
- 52 Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer 2017; 3 (01) 39-55
- 53 Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer 2022; 22 (06) 356-372
- 54 Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun (Camb) 2019; 55 (18) 2704-2707
- 55 Rana S, Bendjennat M, Kour S. et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett 2019; 29 (11) 1375-1379
- 56 Jiang B, Wang ES, Donovan KA. et al. Development of dual and selective degraders of Cyclin-dependent kinases 4 and 6. Angew Chem Int Ed Engl 2019; 58 (19) 6321-6326
- 57 Su S, Yang Z, Gao H. et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem 2019; 62 (16) 7575-7582
- 58 De Dominici M, Porazzi P, Xiao Y. et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020; 135 (18) 1560-1573
- 59 He H, Zhang X, Wang J. et al. Development of degraders of cyclin-dependent kinases 4 and 6 based on rational drug design. J Med Chem 2024; 67 (13) 11354-11364
- 60 Anderson NA, Cryan J, Ahmed A. et al. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg Med Chem Lett 2020; 30 (09) 127106
- 61 Steinebach C, Ng YLD, Sosič I. et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem Sci (Camb) 2020; 11 (13) 3474-3486
- 62 Pu C, Liu Y, Deng R. et al. Development of PROTAC degrader probe of CDK4/6 based on DCAF16. Bioorg Chem 2023; 138: 106637
- 63 Verano AL, You I, Donovan KA. et al. Redirecting the neo-substrate specificity of cereblon-targeting PROTACs to helios. ACS Chem Biol 2022; 17 (09) 2404-2410
- 64 Xiong Y, Zhong Y, Yim H. et al. Bridged proteolysis targeting chimera (PROTAC) enables degradation of undruggable targets. J Am Chem Soc 2022; 144 (49) 22622-22632
- 65 Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 2005; 118 (Pt 22): 5171-5180
- 66 Fisher RP. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019; 10 (02) 47-56
- 67 Larochelle S, Merrick KA, Terret ME. et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell 2007; 25 (06) 839-850
- 68 Schachter MM, Merrick KA, Larochelle S. et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell 2013; 50 (02) 250-260
- 69 Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011; 10 (07) 714-721
- 70 Menzl I, Witalisz-Siepracka A, Sexl V. CDK8-novel therapeutic opportunities. Pharmaceuticals (Basel) 2019; 12 (02) 92
- 71 Lv X, Tian Y, Li S. et al. Discovery and development of cyclin-dependent kinase 8 inhibitors. Curr Med Chem 2020; 27 (32) 5429-5443
- 72 Philip S, Kumarasiri M, Teo T, Yu M, Wang S. Cyclin-dependent kinase 8: a new hope in targeted cancer therapy? Miniperspective. J Med Chem 2018; 61 (12) 5073-5092
- 73 Fant CB, Taatjes DJ. Regulatory functions of the mediator kinases CDK8 and CDK19. Transcription 2019; 10 (02) 76-90
- 74 Xi M, Chen T, Wu C. et al. CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur J Med Chem 2019; 164: 77-91
- 75 Ji W, Du G, Jiang J. et al. Discovery of bivalent small molecule degraders of cyclin-dependent kinase 7 (CDK7). Eur J Med Chem 2024; 276: 116613
- 76 Hatcher JM, Wang ES, Johannessen L, Kwiatkowski N, Sim T, Gray NS. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett 2018; 9 (06) 540-545
- 77 Anshabo AT, Milne R, Wang S, Albrecht H. CDK9: a comprehensive review of its biology, and its role as a potential target for anti-cancer agents. Front Oncol 2021; 11: 678559
- 78 Shen YL, Wang YM, Zhang YX. et al. Targeting cyclin-dependent kinase 9 in cancer therapy. Acta Pharmacol Sin 2022; 43 (07) 1633-1645
- 79 Wu T, Qin Z, Tian Y. et al. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J Med Chem 2020; 63 (22) 13228-13257
- 80 Wu T, Wu X, Xu Y. et al. A patent review of selective CDK9 inhibitors in treating cancer. Expert Opin Ther Pat 2023; 33 (04) 309-322
- 81 Robb CM, Contreras JI, Kour S. et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 2017; 53 (54) 7577-7580
- 82 King HM, Rana S, Kubica SP. et al. Aminopyrazole based CDK9 PROTAC sensitizes pancreatic cancer cells to venetoclax. Bioorg Med Chem Lett 2021; 43: 128061
- 83 Olson CM, Jiang B, Erb MA. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol 2018; 14 (02) 163-170
- 84 Pei J, Xiao Y, Liu X. et al. Piperlongumine conjugates induce targeted protein degradation. Cell Chem Biol 2023; 30 (02) 203-213.e17
- 85 Bian J, Ren J, Li Y. et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem 2018; 81: 373-381
- 86 Qiu X, Li Y, Yu B. et al. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur J Med Chem 2021; 211: 113091
- 87 Wei D, Wang H, Zeng Q. et al. Discovery of potent and selective CDK9 degraders for targeting transcription regulation in triple-negative breast cancer. J Med Chem 2021; 64 (19) 14822-14847
- 88 Tokarski II RJ, Sharpe CM, Huntsman AC. et al. Bifunctional degraders of cyclin dependent kinase 9 (CDK9): probing the relationship between linker length, properties, and selective protein degradation. Eur J Med Chem 2023; 254: 115342
- 89 Chilà R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev 2016; 50: 83-88
- 90 Yan Z, Du Y, Zhang H. et al. Research progress of anticancer drugs targeting CDK12. RSC Med Chem 2023; 14 (09) 1629-1644
- 91 Liu H, Liu K, Dong Z. Targeting CDK12 for cancer therapy: function, mechanism, and drug discovery. Cancer Res 2021; 81 (01) 18-26
- 92 Choi SH, Kim S, Jones KA. Gene expression regulation by CDK12: a versatile kinase in cancer with functions beyond CTD phosphorylation. Exp Mol Med 2020; 52 (05) 762-771
- 93 Paculová H, Kohoutek J. The emerging roles of CDK12 in tumorigenesis. Cell Div 2017; 12: 7
- 94 Liang S, Hu L, Wu Z. et al. CDK12: a potent target and biomarker for human cancer therapy. Cells 2020; 9 (06) 1483
- 95 Tadesse S, Duckett DR, Monastyrskyi A. The promise and current status of CDK12/13 inhibition for the treatment of cancer. Future Med Chem 2021; 13 (02) 117-141
- 96 Jiang B, Gao Y, Che J. et al. Discovery and resistance mechanism of a selective CDK12 degrader. Nat Chem Biol 2021; 17 (06) 675-683
- 97 Niu T, Li K, Jiang L. et al. Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. Eur J Med Chem 2022; 228: 114012
- 98 Yang J, Chang Y, Tien JC. et al. Discovery of a highly potent and selective dual PROTAC degrader of CDK12 and CDK13. J Med Chem 2022; 65 (16) 11066-11083
- 99 Zhou L, Zhou K, Chang Y. et al. Discovery of ZLC491 as a potent, selective, and orally bioavailable CDK12/13 PROTAC degrader. J Med Chem 2024; 67 (20) 18247-18264
- 100 Chang Y, Wang X, Yang J. et al. Development of an orally bioavailable CDK12/13 degrader and induction of synthetic lethality with AKT pathway inhibition. Cell Rep Med 2024; 5 (10) 101752
- 101 Cheng W, Yang Z, Wang S. et al. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur J Med Chem 2019; 164: 615-639
- 102 Hardcastle IR, Golding BT, Griffin RJ. Designing inhibitors of cyclin-dependent kinases. Annu Rev Pharmacol Toxicol 2002; 42: 325-348
- 103 Teng M, Jiang J, He Z. et al. Development of CDK2 and CDK5 dual degrader TMX-2172. Angew Chem Int Ed Engl 2020; 59 (33) 13865-13870
- 104 Zhou F, Chen L, Cao C. et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem 2020; 187: 111952
- 105 Wei M, Zhao R, Cao Y. et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo . Eur J Med Chem 2021; 209: 112903
- 106 Dong G, Ding Y, He S, Sheng C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J Med Chem 2021; 64 (15) 10606-10620
- 107 Zhao L, Zhao J, Zhong K, Tong A, Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther 2022; 7 (01) 113
- 108 Wu H, Yao H, He C. et al. Molecular glues modulate protein functions by inducing protein aggregation: a promising therapeutic strategy of small molecules for disease treatment. Acta Pharm Sin B 2022; 12 (09) 3548-3566
- 109 Fang Y, He Q, Cao J. Targeted protein degradation and regulation with molecular glue: past and recent discoveries. Curr Med Chem 2022; 29 (14) 2490-2503
- 110 Domostegui A, Nieto-Barrado L, Perez-Lopez C, Mayor-Ruiz C. Chasing molecular glue degraders: screening approaches. Chem Soc Rev 2022; 51 (13) 5498-5517
- 111 Toriki ES, Papatzimas JW, Nishikawa K. et al. Rational chemical design of molecular glue degraders. ACS Cent Sci 2023; 9 (05) 915-926
- 112 Słabicki M, Kozicka Z, Petzold G. et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020; 585 (7824) 293-297
- 113 Lv L, Chen P, Cao L. et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 2020; 9: e59994
- 114 Dieter SM, Siegl C, Codó PL. et al. Degradation of CCNK/CDK12 is a druggable vulnerability of colorectal cancer. Cell Rep 2021; 36 (03) 109394
- 115 Jorda R, Havlíček L, Peřina M. et al. 3,5,7-substituted pyrazolo[4,3-d]pyrimidine inhibitors of cyclin-dependent kinases and Cyclin K degraders. J Med Chem 2022; 65 (13) 8881-8896
- 116 Houles T, Boucher J, Lavoie G. et al. The CDK12 inhibitor SR-4835 functions as a molecular glue that promotes cyclin K degradation in melanoma. Cell Death Discov 2023; 9 (01) 459
- 117 Thomas KL, Bouguenina H, Miller DSJ. et al. Degradation by design: new Cyclin K degraders from old CDK inhibitors. ACS Chem Biol 2024; 19 (01) 173-184
- 118 Peng X, Hu Z, Zeng L. et al. Overview of epigenetic degraders based on PROTAC, molecular glue, and hydrophobic tagging technologies. Acta Pharm Sin B 2024; 14 (02) 533-578
- 119 Ha S, Luo G, Xiang H. A comprehensive overview of small-molecule androgen receptor degraders: recent progress and future perspectives. J Med Chem 2022; 65 (24) 16128-16154
- 120 Kastl JM, Davies G, Godsman E, Holdgate GA. Small-molecule degraders beyond PROTACs challenges and opportunities. SLAS Discov 2021; 26 (04) 524-533
- 121 Xie S, Zhu J, Li J. et al. Small-molecule hydrophobic tagging: a promising strategy of druglike technology for targeted protein degradation. J Med Chem 2023; 66 (16) 10917-10933
- 122 He Q, Zhao X, Wu D. et al. Hydrophobic tag-based protein degradation: development, opportunity and challenge. Eur J Med Chem 2023; 260: 115741
- 123 Qiu J, Bai X, Zhang W. et al. LPM3770277, a potent novel CDK4/6 degrader, exerts antitumor effect against triple-negative breast cancer. Front Pharmacol 2022; 13: 853993
- 124 Wang M, Lin R, Li J. et al. Discovery of LL-K8–22: a selective, durable, and small-molecule degrader of the CDK8-cyclin C complex. J Med Chem 2023; 66 (07) 4932-4951
- 125 Li J, Liu T, Song Y. et al. Discovery of small-molecule degraders of the CDK9-Cyclin T1 complex for targeting transcriptional addiction in prostate cancer. J Med Chem 2022; 65 (16) 11034-11057
- 126 Lin R, Yang J, Liu T. et al. Discovery of HyT-based degraders of CDK9-Cyclin T1 complex. Chem Biodivers 2023; 20 (08) e202300769
- 127 Zhong Y, Xu J, Cao H. et al. First ATG101-recruiting small molecule degrader for selective CDK9 degradation via autophagy-lysosome pathway. Acta Pharm Sin B 2025; 15 (05) 2612-2624
- 128 Chen C, Yang Y, Wang Z, Li H, Dong C, Zhang X. Recent advances in Pro-PROTAC development to address on-target off-tumor toxicity. J Med Chem 2023; 66 (13) 8428-8440
- 129 Kounde CS, Shchepinova MM, Saunders CN. et al. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem Commun (Camb) 2020; 56 (41) 5532-5535
- 130 Cheng W, Li S, Wen X. et al. Development of hypoxia-activated PROTAC exerting a more potent effect in tumor hypoxia than in normoxia. Chem Commun (Camb) 2021; 57 (95) 12852-12855
- 131 Shi S, Du Y, Zou Y. et al. Rational design for nitroreductase (NTR)-responsive proteolysis targeting chimeras (PROTACs) selectively targeting tumor tissues. J Med Chem 2022; 65 (06) 5057-5071
- 132 Reynders M, Matsuura BS, Bérouti M. et al. PHOTACs enable optical control of protein degradation. Sci Adv 2020; 6 (08) eaay5064
- 133 Pfaff P, Samarasinghe KTG, Crews CM, Carreira EM. Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent Sci 2019; 5 (10) 1682-1690