Subscribe to RSS
DOI: 10.1055/a-2741-9575
Synthesis, Anticancer Screening, and Virtual Analysis of 5-S-Substituted Derivatives of 1,3-Oxazol-4-ylphosphonates and 1,3-Oxazole-4-carbonitriles
Authors
This work was supported by the National Academy of Sciences of Ukraine under Grants of the NAS of Ukraine to research groups of young scientists of the NAS of Ukraine in 2025-2026 ‘Design, synthesis, in silico and in vitro studies of azole derivatives as potential anticancer agents’ (Contract No. 21/02-2025(6) from 03.03.2025).

Abstract
Ten 5-(arylsulfanyl), 5-(arylsulfinyl), and 5-(arylsulfonyl) derivatives of 1,3-oxazol-4-ylphosphonates and 1,3-oxazole-4-carbonitriles were synthesized and characterized using IR, 1H NMR, 13C NMR, 31P NMR spectroscopy, elemental analysis, and mass spectrometry. Their anticancer activity was assessed against NCI-60 human tumor cell lines using a single-dose assay. Diethyl [2-phenyl-5-(phenylsulfonyl)-1,3-oxazol-4-yl]phosphonate, diethyl [5-(4-chlorophenylsulfonyl)-2-(4-methylphenyl)-1,3-oxazol-4-yl]phosphonate, and 5-(4-methylphenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile, which demonstrated the highest anticancer activity, were selected for five-dose screening. The two phosphonates showed selectivity (SIr > 3 by TGI and LC50) against most leukemia lines, while the 1,3-oxazole-4-carbonitrile derivative was selective against renal (63%), colon (57%), and breast cancer (50%). One compound demonstrated selectivity against the entire leukemia subpanel. A comparison analysis revealed that no standard drug exhibited a high degree of similarity to compounds across all potency vectors. This suggests that the molecular mechanisms may be unique. Possible targets such as cannabinoid receptor 2, adenosine A3 receptor, and cyclin-dependent kinase 2 have been proposed based on in silico studies. The parameters of druglikeness predicted by the ADMET analysis were within the rational range for all compounds. The two phosphonates are expected to be preferable for the development of antileukemia agents, while 5-(4-methylphenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile is considered promising for agents targeting renal, colon, and breast cancers.
Key words
1,3-oxazol-4-ylphosphonates - 1,3-oxazole-4-carbonitriles - synthesis - anticancer screening - COMPARE correlation - ADMET analysis - dockingThe 1,3-oxazole scaffold is widely used in the synthesis of new compounds for various medical purposes. Its derivatives exhibit a variety of pharmacological properties, including anticancer activity, that depend on the presence of substituent groups in the oxazole motif.[1] [2] [3] [4] According to in vitro and ATS studies, the anticancer activity of functionalized oxazoles is due to their interaction with multiple molecular targets involved in the proliferation of cancer cells (e.g., tubulin, DNA topoisomerases, protein kinases, mitochondrial enzymes, aromatase, the estrogen receptor, HDAC, LSD1, HPV E2 TAD, NQO1, Bcl-6, GRP78, STAT3, G-quadruplexes, and the Keap-Nrf2 pathway), thereby reducing growth activity and often inducing apoptosis in cancer cells.[5] Several heterocyclic compounds functionalized with cyano groups have been designed and synthesized as epidermal growth factor receptor (EGFR) inhibitors. These compounds have been shown to inhibit the in vitro growth of various cancer cell lines.[6] [7] [8] [9] [10]
We previously synthesized derivatives of 4-cyano-1,3-oxazole, which exhibited a broad spectrum of antitumor activity against the NCI-60 cancer cell line panel.[11] [12] [13] [14] Based on the results obtained in these studies, further functionalization of 4-cyano-1,3-oxazole was carried out to obtain derivatives with improved anticancer properties.
Diethyl phosphate is known to interact with cardiolipin in biological membranes to form the high-energy ketone cardiolipin enol phosphate in vivo.[15] Cardiolipin plays an important role in stabilizing the mitochondrial electron transport chain complexes. It is involved in cellular signaling associated with oxidative stress, the disruption of which leads to apoptosis.[16] Mitochondria are known to be one of the main targets in the development of anticancer agents, which are designed to attack and destroy cancerous cells.[17] Previously, we synthesized 1,3-oxazol-4-ylphosphonium salts that demonstrated significant anticancer activity against the NCI-60 panel, but with low selectivity.[18] Therefore, in this work, 1,3-oxazol-4-ylphosphonates substituted with diethyl phosphonate were synthesized in the hope of obtaining compounds with improved relative selectivity compared to cancer cells.
In this work, the design, synthesis, antitumor screening, and virtual analysis of 5-(arylsulfanyl), 5-(arylsulfinyl), and 5-(arylsulfonyl) derivatives of 1,3-oxazol-4-ylphosphonates and 1,3-oxazole-4-carbonitriles (Table [1]) were carried out to identify promising new candidates for the development of anticancer agents.
Synthesis
In order to synthesize 4-phosphorylated 5-mercapto-1,3-oxazole derivatives 1–5, a convenient preparative approach was used. It consists of the interaction of phosphorylated 2,2-bis(arylsulfanyl)enamides I obtained from the previously described diethyl esters of 1-(acylamino)-2,2-dichlorovinylphosphonic acids.[19] Enamides I are capable of undergoing intramolecular cyclization under the action of silver carbonate with the formation of 4-phosphorylated 5-mercapto-1,3-oxazole derivatives, including compounds 1 and 2 (Scheme [1]). Reaction of compound 1 with an equivalent of m-chloroperbenzoic acid gives sulfoxide 3. Treatment of compounds 1 and 2 with excess 35% hydrogen peroxide in acetic acid leads to the formation of the corresponding sulfones 4 and 5.[19]
The structure of the synthesized compounds has been reliably proven using 1H, 13C, and 31P NMR spectroscopy and chromatography-mass spectrometry. Thus, the values of the proton signals in the 1H NMR spectra of compounds 1–5 fully correspond to the presented structures. The data of the 13C NMR spectra, in particular, the signals of the carbon nuclei of the oxazole cycle and the ethoxy groups of products 1–5, which appear in the form of doublets due to their interaction with the phosphorus atom nucleus, attract special attention. Thus, in phosphonates 1–5, the signals of C(2) of the oxazole cycle appear in the region of δ = 163.8–162.8. with a spin-spin interaction constant J = 23.0–21.2 Hz and for C(4) in the region of δ = 137.0–133.7. and with J = 238.2–232.5 Hz. It is worth noting that the signals of C(5) in sulfanyl derivatives 1 and 2 are located in the region δ = 148.5–145.1 with J = 36.8–36.7 Hz, and in sulfoxide 3 and sulfones 4 and 5 they are shifted to the region δ = 157.2–151.1 and have J = 36.1–33.5 Hz. The signals of the methylene group of the diethoxyphosphoryl fragment are located at δ = 64.0–62.7 with J = 6.0–5.0 Hz, and the methyl group at δ = 16.5–16.0 with J = 6.5–5.9 Hz.
The signals of the phosphorus nuclei in sulfonyl derivatives 1 and 2 are located in the region of δ = 6.3–6.1, in sulfoxide 3 at δ = 4.4, and in sulfones 4 and 5 at δ = 2.9.
In the IR spectra of compounds 1–5, intense absorption bands of P=O are observed in the region of 1274–1252 cm–1, as well as P–O–C in the region of 1023–1013 cm–1 and 978–973 cm–1. In the IR spectra of sulfones 4 and 5, absorption bands of the O=S=O group are observed at 1350–1344 cm–1 and 1152–1150 cm–1.

2-(Acylamino)-3,3-bis(arylsulfanyl)acrylonitriles II [20] were used to obtain 5-sulfanyl- and 5-sulfonyl-derivatives of 4-cyano-1,3-oxazoles[20] (compounds 6–10) (see Scheme S1 in the Supporting Information).
Biological Testing
One-Dose Assay
The one-dose screening data against the cell lines of the total panel, with percentage growth inhibition (GI) of over 70%, are shown in Table S1. In general, 4-phosphorylated 2-phenyl-1,3-oxazole derivatives 1 (mean GI of 2.8%) and 2 (11.2%), as well as and 4-cyano-2-phenyl-1,3-oxazole derivatives 6 (1.8%), 7 (4.9%), and 8 (2.17%), bearing an arylsulfanyl group at C(5) of the oxazole ring, showed very modest activity against cell lines. Similarly, 5-(4-chlorophenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile (10) demonstrated low activity with a GI of 3.1%. Considering the effect on each cell line individually, compounds 1 and 8 were most effective against the MOLT-4 cell line, with a GI of 32% and 31%, respectively. Compound 2 showed weak activity against the leukemia cell lines K-562 (39%), HS 578T (48%), MOLT-4 (48%), and T-47D (50%), and moderate activity against RPMI-8226 (52%). The most pronounced inhibitory effect of compounds 6 and 7 was observed against the HOP-92 cell line, with a GI of 31% and 34%. The HL-60(BT) cell line was the most sensitive to compound 10, with a GI of 56.6%.
Diethyl 2-phenyl-1,3-oxazol-4-ylphosphonate derivative 3, bearing a phenylsulfinyl group at C(5) of the oxazole ring, and diethyl 2-aryl-1,3-oxazol-4-ylphosphonates 4 and 5 and 2-phenyl-1,3-oxazole-4-carbonitrile 9, containing an arylsulfonyl group at the same position, exhibited the most pronounced anticancer activity among the compounds studied. Compound 3 (mean GI = 38.0%) showed high and very high cytostatic activity against individual cell lines in five subpanels. Compound 9 (36.4%) exhibited selective (SIr > 3) cytotoxicity against seven of twelve sensitive cell lines in six subpanels. The mean inhibitory activity of compound 5 (129.9 ± 4.9%, n = 48) against highly sensitive cell lines (GI ≥ 70%) was slightly, albeit significantly (p = 0.008), higher than that of compound 4 (104.3 ± 5.7%, n = 42). Based on the results of the one-dose screening, compounds 4, 5, and 9 were subjected to a five-dose assay.
Among 1,3-oxazol-4-ylphosphonates 1–5, the introduction of a chlorine atom at the p-position of the phenylsulfanyl group in compound 1 slightly enhanced the activity of the resulting derivative 2. Substitution of the weak electron-donating 4-chlorophenylsulfanyl group in compound 2 with a moderate electron-withdrawing phenylsulfinyl, a strong electron-withdrawing phenylsulfonyl group, or an even stronger electron-withdrawing 4-chlorophenylsulfonyl group led to derivatives with significant antiproliferative (compound 3) and cytotoxic (compounds 4 and 5) activity. This agrees with studies showing that 1,3-oxazole derivatives bearing acceptor substituents at positions 4 and 5 exhibit better inhibition of cancer cell growth than compounds with donor substituents at position 5.[21] [22] The topological indices derived from the graph representation of a molecule and descriptors reflecting its physicochemical characteristics can be used to explain cancer cell growth inhibition. According to the MarvinView data, the tested compounds (ranked by activity rank in relative terms >/<) showed a high and biologically significant positive correlation with the topological index of Wiener polarity, and the physicochemical descriptors listed in Table [2].
a Compound activity rank matches the compound number. PCC = Pearson correlation coefficient. Wp(G) is the Wiener polarity index. Denoted by it is defined as the number of unordered pairs of vertices that are at distance 3 in a molecular graph (the number of 3 bond length distances in the molecule).[23] The WSA (Van der Waals molecular surface area) is defined as the surface of all atomic spheres that are not enclosed by neighboring atomic spheres, depending on their relative orientation. It is the surface through which molecules can interact with each other, keeping them close together.[24] Dreiding energy (DE) is the lowest energy of the 3D structure of the molecule calculated using the Dreiding force field.
The difference in the Wiener polarity index of the analyzed structures between potent active and low active compounds, in this case, reflects only a qualitative relationship between the activity of diethyl 1,3-oxazol-4-ylphosphonate derivatives and the Wiener polarity index, possibly reflecting factors that favor the interaction of the compounds with the target. However, molecular energy, along with the Van der Waals molecular surface area and polar surface area, also gives an idea of the potential of the compound regarding passive permeability through biological membranes, predicting what energy is required for the molecule to pass through the membrane. Therefore, the identified high correlation between these descriptors and the ranked activity, apparently, to a greater extent reflects the influence of the latter factor on the activity of the compounds, since the compounds ranked by the absolute value of activity showed only a moderate and insignificant correlation with these parameters (r < 0.7, p > 0.14).
Among the 2-aryl-1,3-oxazole-4-carbonitrile derivatives, compounds with an arylsulfanyl group (compounds 6–8) showed no significant cancer cell growth inhibition activity. Replacement of the 4-chlorophenylsulfanyl group at position 5 of the 1,3-oxazole ring in the structure of compound 6 by a 4-chlorophenylsulfonyl group gave compound 10 with the same activity. However, substitution of the chlorine atom in the 4-chlorophenylsulfonyl motif of compound 10 by a methyl atom gave compound 9 with better anticancer activity. This compound was weakly active against the entire panel, but showed selective cytostatic activity against individual strains of six subpanels (discussed below in One-Dose Assay).
Five-Dose Assay
Potency
Overall, the differences in the calculated activity parameter values between compounds 4, 5, and 9 did not exceed 40%, suggesting that they are essentially equivalent in their activity about the total panel (Table [3]).
Against five subpanels (non-small lung cancer, melanoma, ovarian cancer, and prostate cancer), the tested compounds showed identical potency in all calculated parameters (p < 0.05). Regarding the remaining sub-panels, compounds 4 and 5 exhibited the greatest antiproliferative (TGI) and cytotoxic (LC50) potencies against leukemia. These were significantly (p < 0.05) 2–5 times higher than those observed for the other subpanels. Compound 5 showed significantly higher GI50 potency against the CNS subpanel, which was three times higher than that of compounds 4 and 9. Compound 9 exceeded the LC50 potency of compound 4 by 2-fold against renal cancer, and compounds 5 and 9 exceeded the LC50 potency of compound 4 by 1.9- and 1.7-fold, respectively, against breast cancer (Table [3]).
For anticancer agents, selectivity for normal cells, along with activity, is the leading indicator of their potential for further studies. However, in the absence of such data, relative selectivity for other cancer cell lines may indirectly reflect the likelihood of side effects due to high genotypic similarity between malignant and normal progenitor cells. None of the compounds showed selectivity for the subpanels tested, except 5, which demonstrated selectivity for leukemia (SIr > 3). However, they selectively inhibited the growth of individual cell lines in other subpanels relative to the overall panel. Except leukemia (against which compounds 4 and 5 demonstrated the highest selectivity), the tested compounds can be ranked in the following order (number of cell lines is given in brackets): TGI: 9 (20) > 5 (7) > 4 (6); LC50: 9 (17) > 5 (9) > 4 (2). In addition, compound 9 selectively inhibited the growth of five out of eight (63%) renal cancer cell lines, four out of seven (57%) colon cancer cell lines by both parameters, and 50% of breast cancer cell lines by TGI (Table [3]). Hence, compounds 4 and 5 are promising leads for further functionalization and development as antileukemic agents, and compound 9 against renal cancer, colon cancer, and breast cancer.
a The value of the relative selectivity index is given in brackets, allowing the compound to be classified as acting selectively (SIr > 3). The selectivity on the GI50 vector was not calculated.
COMPARE Correlation
Regarding the antiproliferative activity vectors (GI50 and TGI), compound 9 exhibited only moderate correlation with rifamycin SV (0.57 and 0.59, respectively). No standard drug showed even a moderate correlation (r > 0.5) for cytotoxicity, demonstrating only a weak correlation with rhizoxin (r = 0.4). The low correlation of compound 9 with standard drugs across all potency vectors suggests the uniqueness of its anticancer activity. Unfortunately, COMPARE analyses of compounds 4 and 5 were unable to generate results.
In Silico Prediction of Possible Molecular Targets
The ability to selectively bind to and modulate the intended molecular target is an important aspect in the mechanism of action of a drug. Therefore, identifying probable molecular targets for bioactive compounds and describing the likelihood of their interaction is an integral component at the early stages of drug development.[25]
The SwissTargetPrediction web server, which proposes potential targets based on 2D and 3D structural similarity between studied compounds and ligands of known biomolecules, was used to predict the molecular targets of the 5-arylsulfanyl-substituted derivatives of 1,3-oxazol-4-ylphosphonates and 1,3-oxazole-4-carbonitriles. According to the obtained results, cannabinoid receptor 2 can be the target for compounds 4 and 5, and serotonin 5-HT6 receptor and adenosine receptors are proposed as targets for compound 9. This correlates with anticancer screening data, as is most clearly manifested in the case of leukemia. The cannabinoid receptor 2 is expressed in the immune system, including in B cells, T cells, and monocytes/macrophages.[26] In experimental and clinical studies, the cannabinoid receptor 2 agonists have been shown to induce apoptosis in leukemia cells, but not in normal human peripheral blood lymphocytes.[27] [28] The antipsychotic drug sertindole, acting as a 5-HT6 receptor antagonist, has demonstrated antiproliferative effects in various cancer modes, particularly against breast and leukemia cancer cell lines.[29] The adenosine A3 receptor is highly expressed in various tumor cells, and its agonists have been shown to induce tumor growth inhibition.[30]
5-Arylsulfanyl-substituted derivatives of 1,3-oxazol-4-ylphosphonates 4 and 5, as well as 5-(4-methylphenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile (9), were docked into the orthosteric sites of the cannabinoid receptor 2, serotonin 5-HT6 receptor, and adenosine A3 receptor. In addition to these potential targets, the compounds were also evaluated as promising ligands for the colchicine-binding site of tubulin and the ATP-binding sites of BRAF V600E mutant and CDK2. It is well known that microtubule assembly plays a key role in essential cellular processes, and its disruption by specific inhibitors is considered a promising strategy in anticancer therapy.[31] In turn, BRAF is a component of the key intracellular RAS/RAF/MEK/ERK signaling cascade, which regulates cellular proliferation, differentiation, growth, and survival, whereas CDK2 controls cell cycle progression. Therefore, these protein kinases represent attractive targets for the development of anticancer drugs.[32] [33]
As shown by the docking energies presented in Table [4], compounds 4, 5, and 9 exhibit significant affinities for the cannabinoid binding site of CB2, supporting the prediction made by the SwissTargetPrediction web server. In addition, compound 4 demonstrated comparable docking energy for the ATP-binding site of CDK2. In contrast, compound 9 exhibited a similar binding score for the adenosine-binding site of A3AR and the ATP-binding sites of BRAF and CDK2.
The binding modes shown in Figure [1] suggest that diethyl [5-(4-chlorophenylsulfonyl)-2-(4-methylphenyl)-1,3-oxazol-4-yl]phosphonate (5) and 5-(4-methylphenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile (9) occupy the cannabinoid binding site of CB2 and the adenosine-binding site of A3AR, respectively, forming multiple hydrophobic interactions with the surrounding hydrophobic amino acid residues.

In addition, the oxazole ring of compound 5 engages in a π-stacking interaction with Phe183, while the diethyl phosphonate group forms a π-cation interaction with Phe117 and a hydrogen bond with Ser285. In the case of compound 9 binding to the adenosine-binding site of A3AR, the oxazole ring, together with the 4-methylphenylsulfonyl moiety at position 5, forms π-stacking interactions with Phe168. One of the oxygen atoms of the sulfonyl group is involved in a hydrogen bond with Tyr15, while the cyano group forms similar interactions with Gln167 and Tyr265.
Passing the tested compounds through druglikeness filters, compounds 4 and 9 passed all filters used for assessing bioavailability (see Table [5]). Compound 5 also passed most of these filters, except the Ghose and Egan rules (the LogP threshold was exceeded). However, depending on the method used, the SwissADME calculated LogP values for compound 5 vary widely, from 2.61 to 6.11, with a consensus value of 4.18. Therefore, this parameter requires experimental determination and is not critical in this case.
Compound 9 also passed all filters, removing molecules with undesirable properties. However, compounds 4 and 5 were rejected by the Brenk filter due to the presence of phosphorus, suggesting their poor pharmacokinetics (Table [6]), which will be predicted below.
a PAINS = criterion for excluding compounds that interfere with the analysis. Brenk is used to filter out compounds that have poor pharmacokinetics. BMS = the Bristol-Myers Squibb rule is used to filter undesirable reactive compounds that could cause severe toxicities. Synthetic availability index ≤ 6 is an indication that the compound is readily synthesizable.
ADMET Analysis
Virtual drug similarity analysis is designed to determine the potential of a molecule as a drug, in order to eliminate compounds likely to fail in clinical trials at an early stage. ADMET filters are used to estimate the probability that a chemical substance will exhibit specific pharmacological activity with minimal side effects. Conversely, passing several ADMET filters increases the likelihood that a substance under development will become a drug candidate.[34] Due to the impossibility of making a confident conclusion, the discussion of data showing the probability of manifestation of properties of compounds in the range 0.3 < p < 0.7 is excluded from the ADMET analysis conducted below.[35]
ADMET analysis enables the prediction of the pharmacokinetic profile of compounds, which is essential for evaluating the pharmacodynamic activity of molecules. The predicted pharmacokinetic properties of the compounds are presented in Table [7].
a Papp = apparent permeability coefficient, Peff = resulting effective permeability, HIA = human intestinal absorption. Molecules with HIA ≥ 30% were classified as absorbable. PPB = plasma protein binding, optimal: < 90%; drugs with high protein-binding may have a low therapeutic index. F50% = human oral bioavailability ≥ 50%. Fu = the fraction unbound in plasmas, low: < 5%. VD = volume distribution, optimal range: 0.04–20 L/kg; a high VD value indicates that the drug is highly distributed to the tissues, while a low VD value indicates that the drug is primarily confined to the plasma. CL = plasma clearance, low: < 5 mL/min/kg.
b Probabilistic assessment (0 ≥ p ≤ 1) if units of measurement are not specified.
In general, all tested compounds are predicted to have acceptable absorption properties for drug candidates. The same applies to the estimated distribution parameters, except PPB and Fu, suggesting that they will show a low therapeutic index in clinical use. The predicted absence of substrate binding indicates Pgp-independent transport of these compounds across cell membranes. Their inhibition of Pgp expressed on apical cell membranes promotes increased bioavailability for other drugs that are its substrate, increasing their systemic exposure.[36]
The overexpression of Pgp, MRP1, and BCRP (which are efflux transporters that use ATP energy to expel anticancer drugs from cells against a concentration gradient) leads to multidrug resistance in cancer cells.[37] MRP1 is a basolateral transporter that moves compounds into tissues located beneath the basement membrane. It therefore plays an important role in the functioning of blood barriers by preventing drugs from reaching their post-barrier targets.[38] All compounds were predicted to exhibit inhibitory activity and lack Pgp substrate binding, indicating a non-competitive (allosteric) mechanism of inhibition. This suggests their potential for use in combination with anticancer agents that have limited efficacy due to the development of resistance during treatment.[39] [40] It is also assumed that the tested compounds are MRP1 inhibitors. In this case, they will promote the penetration of MRP1 substrates through the blood-brain, blood-placenta, blood-retina, blood-testis, and blood-thymus barriers, making the corresponding tumors more accessible to anticancer drugs that do not penetrate through blood barriers when used together.
The cytochrome P450 (CYP450) enzyme family plays a significant role in xenobiotic metabolism. Among these, CYP3A4 is the most significant, metabolizing around 50% of known xenobiotics in humans.[41] The substrate specificity of this cytochrome was predicted for compounds 5 and 9, which may also be substrates for CYP2C19 and CYP2C9. Compound 4, on the other hand, is not expected to be metabolized by any of the listed cytochromes. This means it is not expected to undergo oxidative biotransformation to intermediate metabolites with unpredictable biological activity.
CL and T1/2 are related to the available systemic dose and are important for predicting dosage and dosing intervals. The low CL and T1/2 values of the test compounds suggest that the dosing interval should be increased to achieve steady-state concentrations, since the drug is eliminated from the body slowly and has a short duration of action.
ADMETlab3.0 predicted a low probability of interaction between these compounds and stress signaling pathways (SR) and nuclear receptors (NR), except for NR-aromatase (compound 4) and SR-MMP. For the latter, a high probability of interaction with all tested compounds was predicted for compounds 4 and 5, and a very high probability for compound 9. However, Deep-PK predicts a very low probability of interaction with these targets for all tested compounds, and a high or very high probability for compounds 4 and 5 with SR-ARE. This does not allow us to confidently evaluate the results of virtual forecasting (Table S2).
For normal cells, the test compounds are predicted to have a low probability of causing cardiotoxicity, hematotoxicity, and ototoxicity, but a very high probability of causing genotoxicity. Additionally, unlike compound 9, compounds 4 and 5 may exhibit nephrotoxicity. The predicted probabilities of human hepatotoxicity and respiratory toxicity require further evaluation (see Table S3). In general, given the high cytotoxicity inherent a priori to all chemotherapeutic agents, the predicted in silico results are acceptable.
Conclusion
Antitumor screening and virtual analysis results show that phosphonates 4 and 5, among 5-arylsulfonyl-substituted derivatives of 1,3-oxazol-4-ylphosphonates, and 1,3-oxazole-4-carbonitrile 9, among 5-arylsulfonyl-substituted derivatives of 1,3-oxazole-4-carbonitriles, exhibit high potency against multiple cancer cell lines. Meanwhile, diethyl phosphonate esters 4 and 5 exhibited high selectivity for leukemia. Therefore, these phosphonates are expected to be favored in the development of antileukemia agents. 5-(4-Methylphenylsulfonyl)-2-phenyl-1,3-oxazole-4-carbonitrile (9) is a promising candidate for further study as a potential therapeutic agent against renal, colon, and breast cancers, given its selective toxicity towards these cancer cell lines. Based on in silico studies, it was suggested that the anticancer activity of compounds 4, 5, and 9 may be realized through interaction with cannabinoid receptor 2, adenosine A3 receptor, and cyclin-dependent kinase 2.
All reagents and solvents used in synthetic procedures were purchased from Aldrich and used as received. The reactions were followed by TLC (silica gel, aluminum sheets 60 F254, Merck). Melting points were recorded on a Fisher-Johns apparatus. IR spectra were recorded on a Vertex-70 spectrophotometer. The samples were prepared either as KBr pellets or as films. 1H, 13C, and 31P NMR spectra were recorded on a Varian Mercury spectrometer (300 or 400 and 101 MHz, respectively) or Bruker Avance DRX 500 spectrometer (500, 126, and 162 MHz respectively) in CDCl3 or DMSO-d 6, taking its residual solvent signal as a standard. LC-MS analysis was performed on an Agilent 1200 Series system equipped with a diode array and a G6130A mass-spectrometer (atmospheric pressure electrospray ionization). Combustion elemental analysis was performed in the V.P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry analytical laboratory; their results were found to be in good agreement (±0.4%) with the calculated values. The carbon and hydrogen contents were determined using the Pregl gravimetric method, nitrogen (using Dumas’ gasometrical micromethod), sulfur (by the Scheininger titrimetric method), chlorine (by the mercurimetric method), and phosphorus (by the colorimetric method).
The general procedure for the synthesis of 5-S-substituted 1,3-oxazol-4-ylphosphonates 1–5 is provided in the Supporting Information.
Diethyl [2-Phenyl-5-(phenylsulfanyl)-1,3-oxazol-4-yl]phosphonate (1)
Colorless oil; yield: 83%.
IR (film): 1259 (P=O), 1054, 1015 (P–O–C), 973 (P–O–C–C), 746, 689, 600, 536 cm–1.
1H NMR (400 MHz, DMSO-d 6): δ = 7.91 (d, J HH = 6.5 Hz, 2 H, ArH), 7.57–7.33 (m, 8 H, ArH), 4.15–4.12 (m, 4 H, P(OCH 2CH3)2), 1.26 (t, J HH = 7.0 Hz, 6 H, P(OCH2 CH 3)2).
13C NMR (126 MHz, DMSO-d 6): δ = 163.6 (d, J CP = 23.0 Hz), 149.1 (d, J CP = 36.7 Hz), 135.0 (d, J CP = 238.2 Hz), 131.7 131.6, 129.8, 129.7, 129.3, 128.1, 126.4, 125.5, 62.7 (d, J CP = 5.9 Hz), 16.0 (d, J CP = 6.3 Hz).
31P NMR (162 MHz, DMSO-d 6): δ = 6.3.
LC-MS: m/z (%) = 390.0 (100) [M + 1]+.
Anal. Calcd for C19H20NO4PS: C, 58.60; H, 5.18; N, 3.60; P, 7.95; S, 8.23. Found: C, 58.84; H, 5.11; N, 3.91; P, 7.86; S, 8.31.
Diethyl [2-Phenyl-5-(phenylsulfinyl)-1,3-oxazol-4-yl]phosphonate (3)
To a solution of compound 1 (0.01 mol) in CH2Cl2 (50 mL) was added a saturated solution of m-CPBA (0.011 mol) in CH2Cl2 at 10–15 °C. The mixture was stirred at this temperature for 12 h. The precipitate was filtered off; the filtrate was treated with sat. Na2CO3 solution, dried (Na2SO4), and the solvent was removed in vacuo. The oily substance was reprecipitated from petroleum ether as a colorless oil; yield: 83%.
IR (film): 1252 (P=O), 1219 (S=O), 1013 (P–O–C), 978 (P–O–C–C), 752, 688, 594, 508 cm–1.
1H NMR (400 MHz, DMSO-d 6): δ = 7.88 (d, J HH = 8.0 Hz, 2 H, ArH), 7.85 (d, J HH = 8.0 Hz, 2 H, ArH), 7.66–7.54 (m, 6 H, ArH), 4.29–4.13 (m, 4 H, P(OCH 2CH3)2), 1.35–1.27 (m, 6 H, P(OCH 2CH3)2).
13C NMR (126 MHz, DMSO-d 6): δ = 163.7 (d, J CP = 21.2 Hz), 157.2 (d, J CP = 36.1 Hz), 141.0, 134.1 (d, J CP = 233.9 Hz), 132.5 132.0, 129.8, 129.5, 126.8, 124.7, 124.6, 63.4 (d, J CP = 5.0 Hz), 63.3 (d, J CP = 5.0 Hz), 16.1 (d, J CP = 6.0 Hz), 16.0 (d, J CP = 6.0 Hz).
31P NMR (162 MHz, DMSO-d 6): δ = 4.36.
LC-MS: m/z (%) = 406.0 (100) [M + 1]+.
Anal. Calcd for C19H20NO5PS: C, 56.29; H, 4.97; N, 3.45; P, 7.64; S, 7.91. Found: C, 56.01; H, 5.12; N, 3.68; P, 7.69; S, 7.83.
Diethyl Esters of 2-Aryl-5-(arylsulfonyl)-1,3-oxazol-4-ylphosphonic Acids 4 and 5
To a solution of diethyl 2-aryl-5-(arylsulfanyl)-1,3-oxazol-4-ylphosphonic acid (0.01 mol) in glacial AcOH (20 mL), was added 35% aq H2O2 in three 2 mL portions over 2 h. The mixture was kept at 20–25 °C for 8 h, the precipitate was filtered off and purified by crystallization (EtOH).
Diethyl [2-Phenyl-5-(phenylsulfonyl)-1,3-oxazol-4-yl]phosphonate (4)
Colorless solid; yield: 83%; mp 116 °C.
IR (KBr): 1350 (O=S=O), 1267 (P=O), 1180, 1152 (O=S=O), 1059, 1023 (P–O–C), 977 (P–O–C–C), 728, 686, 626, 582, 556 cm–1.
1H NMR (400 MHz, DMSO-d 6): δ = 8.2 (d, J HH = 8.3 Hz, 2 H, ArH), 8.0 (d, J HH = 7.0 Hz, 2 H, ArH), 7.84–7.80 (m, 1 H, ArH), 7.74–7.70 (m, 2 H, ArH), 7.67–7.64 (m, 1 H, ArH), 7.61–7.57 (m, 2 H, ArH), 4.25–4.16 (m, 4 H, P(OCH 2CH3)2), 1.30 (t, J HH = 6.8 Hz, 6 H, P(OCH 2CH3)2).
13C NMR (126 MHz, DMSO-d 6): δ = 162.9 (d, J CP = 22.0 Hz), 151.2 (d, J CP = 33.6 Hz), 138.6, 137.0 (d, J CP = 232.5 Hz), 135.2, 132.7, 129.9, 129.4, 128.1, 127.2, 124.5, 63.5 (d, J CP = 6.0 Hz), 16.1 (d, J CP = 6.5 Hz).
31P NMR (162 MHz, DMSO-d 6): δ = 2.9.
LC-MS: m/z (%) = 422.0 (100) [M + 1]+.
Anal. Calcd for C19H20NO4PS: C, 58.60; H, 5.18; N, 3.60; P, 7.95; S, 8.23. Found: C, 58.79; H, 5.10; N, 3.74; P, 7.83; S, 8.41.
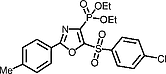
Diethyl [5-(4-Chlorophenylsulfonyl)-2-(4-methylphenyl)-1,3-oxazol-4-yl]phosphonate (5)
Colorless solid; yield: 79%; mp 110 °C.
IR (KBr): 2988, 1612, 1577, 1548, 1490, 1344 (O=S=O), 1274 (P=O), 1221, 1180, 1150 (O=S=O), 1087, 1022 (P–O–C), 977 (P–O–C–C), 957, 829, 758, 654, 615, 578, 546 cm–1.
1H NMR (400 MHz, DMSO-d 6): δ = 8.18 (d, J HH = 8.90 Hz, 2 H, ArH), 7.90 (d, J HH = 8.00 Hz, 2 H, ArH), 7.79 (d, J HH = 8.9 Hz, 2 H, ArH), 7.40 (d, J HH = 8.1 Hz, 2 H, ArH), 4.29–4.12 (m, 4 H, P(OCH2 CH 3)2), 2.38 (s, 3 H), 1.30 (t, J HH = 7.0 Hz, 6 H, P(OCH2 CH 3)2).
13C NMR (126 MHz, DMSO-d 6): δ = 163.7 (d, J CP = 21.9 Hz), 151.1 (d, J CP = 33.5 Hz), 143.7, 140.8, 138.0, 137.0 (d, J CP = 234.8 Hz), 130.6, 130.5, 130.4, 127.8, 122.3, 64.03 (d, J CP = 5.9 Hz), 16.5 (d, J CP = 5.9 Hz).
31P NMR (162 MHz, DMSO-d 6): δ = 2.9.
LC-MS: m/z (%) = 470.0 (100) [M + 1]+.
Anal. Calcd for C20H21ClNO6PS: C, 51.12; H, 4.50; Cl, 7.55; N, 2.98; P, 6.59; S, 6.82. Found: C, 51.29; H, 4.67; Cl, 7.47; N, 3.15; P, 6.47; S, 6.96.
The general procedure for the synthesis 1,3-oxazole-4-carbonitriles 6–10, together with their spectral data, is provided in the Supporting Information.
In Vitro Anticancer Screening of the Tested Compounds
One Dose Assay
Synthesized compounds were submitted to the National Cancer Institute (NCI), Bethesda, Maryland, U.S.A., under the Developmental Therapeutic Program DTP. Primary in vitro one dose anticancer assay was performed using the total cell line panel by the protocol of the Drug Evaluation Branch, NCI, Bethesda.[42] [43] [44] The compounds were added at a single concentration (10 μM), and the culture was incubated for 48 h. End point determinations were made with a protein-binding dye, sulforhodamine B (SRB). Results for each compound were reported as the percent of growth of the treated cells when compared to the untreated control cells. The following criteria were used for the qualitative assessment of the antiproliferative activity of the compounds (GI range: 0–100%): GI < 50%: weak activity; 50% ≤ GI < 70%: moderate activity; 70% ≤ GI < 90%: high activity; 90% ≤ GI: very high activity. Similar criteria were applied to the cytotoxicity parameter (GI range: 101–200%).
Five-Dose Assay
The screening compounds were tested against breast cancer cell lines of the NCI subpanel. Cells of all breast cancer lines were incubated at five different concentrations (0.01, 0.1, 1, 10, and 100 μM) of the tested compounds. The outcomes were used to create three response parameters (GI50, TGI, and LC50), which were calculated for each cell line and each experimental compound. These calculations were made according to the method described by the NCI/NIH Development Therapeutics Program.[45] The GI50 value is a measure of the sensitivity of a cell to the effect of the drug and corresponds to the concentration of the compound causing a 50% decrease in net cell growth. TGI is the concentration of the study drug that causes total inhibition of cell growth. The LC50 value is the concentration of the compound causing a net 50 % loss of initial cells at the end of the incubation period of 48 h.
The GI50 is determined as the drug concentrations that result in 50% and 0% growth at 48 h drug exposure. Growth inhibition was calculated from the equation: [(T – T0)/(C – T0)] × 100 = 50. The TGI, which signifies a cytostatic effect, was calculated from the equation: 100 × (T – T0)/(C – T0) = 0. The LC50, which signifies a cytotoxic effect, was calculated from the equation: [(T – T0)/T0] × 100 = –50, when T < T0. Where: T0 is the cell count at day 0; C is the vehicle control (the cell count without drug at the end of the incubation period), and T is the cell count at the test concentration of drug at the end of the incubation period.
The relative selectivity (SIr ) criterion for identifying prospective anti-cancer compounds is calculated as follows: SIr = PAtotal/PAcell, where PAtotal is the average activity parameter (GI, GI50, TGI, or LC50) of the compound against the total panel of cancer cell lines. PAcell is the corresponding activity parameter of the compound against a particular cancer cell line. SIr ratios of 3–6 indicate selectivity; ratios greater than 6 indicate moderate selectivity towards the corresponding cell line; and compounds with SIr < 3 are considered non-selective.[46] [47]
COMPARE Correlations
The graph of mean values for each compound was subsequently used to run the COMPARE algorithm from the Developmental Therapeutics Program, NCI, and calculate the correlation coefficient concerning compounds from the standard agent database with a known mechanism of action.[48] Pairwise correlation coefficient ≥ 0.70 was used as the cut-off for assessing whether two agents were likely to share a similar mechanism of action. Briefly, vectors of GI50, TGI, and LC50 concentrations for the tested compound were correlated with the set of average GI50, TGI, and LC50 vectors for all public NCI-60 vectors for the full public standard agents database.[49]
Molecular Targets Prediction
The probable molecular targets were identified for the most active anticancer compounds 4, 5, and 9 using the SwissTargetPrediction web server (http://www.swisstargetprediction.ch/).[50] Following these results, molecular docking studies were performed to validate the compounds as potential ligands of the orthosteric sites of predicted targets. Given the potential of 2-phenyl-1,3-oxazole derivatives to influence microtubule assembly,[51] [52] compounds 4, 5, and 9 were also docked to the colchicine-binding site of tubulin. In addition, given that the anticancer activity of oxazole derivatives may involve multiple mechanisms,[5] molecular docking studies was carried out to evaluate the binding interactions of the compounds with the ATP-binding sites of B-Raf kinase domain V600E mutant and cyclin-dependent kinase 2, both of which are well-established anticancer targets.
The crystal structures of cannabinoid receptor 2 (CB2, 6PT0),[53] serotonin 5-HT6 receptor (5-HT6, 7YS6),[54] adenosine A3 receptor (A3AR, 8X16),[55] tubulin (PDB ID: 1SA0),[56] B-Raf kinase domain with oncogenic V600E mutation (BRAF, 4MNF),[57] and cyclin-dependent kinase 2 (CDK2, 3QQJ),[58] were obtained from Protein Data Bank (www.rcsb.org).[59] For molecular docking calculations using AutoDock Vina 1.1.2,[60] the following structures were used: cannabinoid, serotonin, and adenosine receptors; the A and B subunits of tubulin; and the A subunit of BRAF and CDK2 were used. Before performing the computations, ligands, water molecules, and protein subunits not involved in the docking process were removed from the downloaded files employing Discovery Studio 3.5 (Accelrys, San Diego, USA). The structures of compounds 4, 5, and 9 were drawn in Marvin Sketch,[61] saved in MOL2 format, and then optimized using Avogadro[62] with MMFF94s force field. The MOL2 files of ligands were loaded into AutoDockTools 1.5.6[63] and saved in PDBQT format while keeping charges. Each PDB file of the proteins was loaded into AutoDockTools 1.5.6, hydrogen atoms were added, Gasteiger charges were calculated, and the prepared protein structure was saved in PDBQT format. The grids box size was set to 20 × 20 × 20 and grids centers were set to 98.213, 109.469, 124.446 (CB2); 115.317, 177.547, 138.578 (5-HT6); 125.593, 99.797, 80.509 (A3AR); 118.934, 89.717, 5.964 (tubulin); 15.264, –26.772, –3.275 (BRAF); and –11.781, –80.97, –12.377 (CDK2).
To validate the docking protocols, the co-crystallized ligands were prepared and re-docked into their native binding sites. The obtained docking energies were –12.2 kcal/mol for CB2, –6.1 kcal/mol for 5-HT6, –9.2 kcal/mol for A3AR, –8.6 kcal/mol for tubulin, –10.6 kcal/mol for BRAF, and –8.2 kcal/mol for CDK2. The RMSD values (based on heavy atoms) between the docked and crystallographic poses of the co-crystallized ligands, which were 2.0 Å for CB2, 1.6 Å for 5-HT6, 1.7 Å for A3AR, 1.2 Å for tubulin, 0.99 Å for BRAF, and 0.28 Å for CDK2, demonstrate the validity and robustness of the docking protocols.
ADMET Analysis
Available online websites of an integrated online platform for Windows: ADMETlab 3.0,[64] SwissADME,[65] and Deep-PK[66] are applied to explore ADMET properties of the studied molecules. The pharmacokinetic, pharmacodynamic, and toxic targets for the test compounds were predicted. The conversion of molecular structures into SMILES strings, which is required for the operation of ADMET platforms, was done using the Marvin JS widget.[67] The latter platform was also used for the SAR analysis.
Statistical Data Analysis
Statistical analyses of the results were performed using the program Statistica v6.0 for Windows. Statistically significant differences between groups were evaluated using the unpaired Student t-test (p < 0.05). The data are given as means ± SEM (standard error of mean).
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We thank the National Cancer Institute, Bethesda, MD, USA, for the in vitro evaluation of anticancer activity within the Developmental Therapeutic Program and Enamine Ltd for the material and technical support.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/a-2741-9575.
- Supporting Information (PDF) (opens in new window)
-
References
- 1 Kakkar S, Narasimhan B. BMC Chem. 2019; 13: 16
- 2 Joshi S, Mehra M, Singh R, Kakar S. Egypt. J. Basic Appl. Sci. 2023; 10: 218
- 3 Yan X, Jing W, Zhou L, Fan L, Wang X, Xu Z. Curr. Top. Med. Chem. 2020; 20: 1916
- 4 Sawant RS, Deventhiran P, Uppalwar SV, Sen AK. IJRPR 2024; 5: 1510
- 5 Kulkarni S, Kaur K, Jaitak V. Anti-cancer Agents Med. Chem. 2022; 22: 1859
- 6 El-Dydamony NM, Abdelnaby RM, Abdelhady R, Ali O, Fahmy MI, Eldeen R FakhrR, Helwa AA. J. Enzyme Inhib. Med. Chem. 2022; 37: 895
- 7 Reda N, Mohamed KO, Abdou K, Helwa AA, Elshewy A. Bioorg. Chem. 2024; 145: 107185
- 8 Al-Wahaibi LH, Abou-Zied HA, Hisham M, Beshr EA. M, Youssif BG. M, Bräse S, Hayallah AM, Abdel-Aziz M. Molecules 2023; 28: 6586
- 9 Saffari Z, Aryapour H, Akbarzadeh A, Foroumadi A, Jafari N, Farahnak Zarabi M, Farhangi A. Tumour Biol. 2014; 35: 5845
- 10 Ahmed NM, Mohamed MS, Awad SM, Abd El-Hameed RH, El-Tawab NA. A, Gaballah MS, Said AM. J. Enzyme Inhib. Med. Chem. 2024; 39: 2304625
- 11 Kachaeva MV, Pilyo SG, Demydchuk BA, Prokopenko VM, Zhirnov VV, Brovarets VS. Int. J. Curr. Res. 2018; 10: 69410
- 12 Kachaeva MV, Pilyo SG, Zhirnov VV, Brovarets VS. Med. Chem. Res. 2019; 28: 71
- 13 Severin O, Pilyo S, Kachaeva M, Zhirnov V, Hodyna D, Bahrieieva O, Brovarets V. ChemMedChem 2024; 19: e202300527
- 14 Severin O, Pilyo S, Semenyuta I, Kachaeva M, Zhirnov V, Brovarets V. Curr. Chem. Lett. 2025; 14: 159
- 15 Schole J, Schole C, Eikemeyer J. Z. Naturforsch., C 1994; 49: 657
- 16 Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Cells 2019; 8: 728
- 17 Kenny TC, Birsoy K. Cold Spring Harbor Perspect. Med. 2024; 14: a041534
- 18 Brusnakov M, Golovchenko O, Velihina Y, Liavynets O, Zhirnov V, Brovarets V. ChemMedChem 2022; 17: e202200319
- 19 Kondratyuk KM, Lukashuk EI, Golovchenko AV, Vasilenko AN, Brovarets VS. Russ. J. Gen. Chem. 2013; 83: 46
- 20 Pil’o SG, Brovarets VS, Vinogradova TK, Golovchenko AV, Drach BS. Russ. J. Gen. Chem. 2002; 72: 1714
- 21 Kachaeva MV, Hodyna M, Obernikhina NV, Pilyo SG, Kovalenko YS, Prokopenko VM, Kachkovsky OD, Brovarets VS. J. Heterocycl. Chem. 2019; 56: 3122
- 22 Obernikhina NV. OMCIJ 2019; 9: 555751
- 23a Lukovits I, Linert W. J. Chem. Inf. Comput. Sci. 1998; 38: 715
- 23b Chen L, Li T, Liu J, Shi Y, Wang H. PLoS One 2016; 11: e0167075
- 24 Whitley DC. J. Math. Chem. 1998; 23: 377
- 25 Agoni C, Olotu FA, Ramharack P, Soliman ME. J. Mol. Model. 2020; 26: 120
- 26 Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G, Casellas P. Eur. J. Biochem. 1993; 214: 173
- 27 Soto-Mercado V, Mendivil-Perez M, Jimenez-Del-Rio M, Fox JE, Velez-Pardo C. Leuk. Res. 2020; 95: 106389
- 28 Melén CM, Merrien M, Wasik AM, Panagiotidis G, Beck O, Sonnevi K, Junlén HR, Christensson B, Sander B, Wahlin BE. Leuk. Lymphoma 2022; 63: 1387
- 29 Zhang W, Zhang C, Liu F, Mao Y, Xu W, Fan T, Sun Q, He S, Chen Y, Guo W, Tan Y, Jiang Y. Sci. Rep. 2018; 8: 15753
- 30 Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. Immunol., Endocr. Metab. Agents Med. Chem. 2007; 7: 298
- 31 Peerzada MN, Dar MS, Verma S. Expert Opin. Ther. Pat. 2023; 33: 797
- 32 Maji L, Teli G, Raghavendra NM, Sengupta S, Pal R, Ghara A, Matada GS. P. Mol. Diversity 2024; 28: 2689
- 33 Marak BN, Dowarah J, Khiangte L, Singh VP. Eur. J. Med. Chem. 2020; 203: 112571
- 34 Lee K, Jang J, Seo S, Lim J, Kim WY. Chem. Sci. 2021; 13: 554
- 35 Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A, Chen X, Hou T, Cao D. Nucleic Acids Res. 2021; 49: W5
- 36 Patel D, Sethi N, Patel P, Shah S, Patel K. Eur. J. Pharm. Biopharm. 2024; 198: 114267
- 37 Gonçalves BM. F, Cardoso DS. P, Ferreira MJ. U. Molecules 2020; 25: 3364
- 38 Elfadadny A, El-Husseiny HM, Abugomaa A, Ragab RF, Mady EA, Aboubakr M, Samir H, Mandour AS, El-Mleeh A, El-Far AH, Abd El-AzizA. H, Elbadawy M. Environ. Sci. Pollut. Res. Int. 2021; 28: 49447
- 39 Husain A, Makadia V, Valicherla GR, Riyazuddin M, Gayen JR. Drug Dev. Res. 2022; 83: 825
- 40 Tian Y, Lei Y, Wang Y, Lai J, Wang J, Xia F. Int. J. Oncol. 2023; 63: 119
- 41 Guengerich FP. Annu. Rev. Pharmacol. Toxicol. 1999; 39: 1
- 42 Grever MR, Schepartz SA, Chabner BA. Semin. Oncol. 1992; 19: 622
- 43 Boyd MR, Paull KD. Drug Dev. Res. 1995; 34: 91
- 44 Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Jangley J, Cronisie P, Viagro-Wolff A, Gray-Goodrich M, Campell H, Boyd M. J. Natl. Cancer Inst. 1991; 83: 757
- 45 Data (accessed Nov 21, 2025): https://dtp.cancer.gov/discovery_development/nci-60/default.htm
- 46 Acton EM, Narayanan VL, Risbood PA, Shoemaker RH, Vistica DT, Boyd MR. J. Med. Chem. 1994; 37: 2185
- 47 Weerapreeyakul N, Nonpunya A, Barusrux S, Thitimetharoch T, Sripanidkulchai B. Chin. Med. 2012; 7: 15
- 48 History of the NCI-60 screen and COMPARE algorithm (accessed Nov 21, 2025): https://dtp.cancer.gov/databases_tools/compare.htm
- 49 Shoemaker RH. Nat. Rev. Cancer. 2006; 6: 813
- 50 Daina A, Michielin O, Zoete V. Nucleic Acids Res. 2019; 47: W357
- 51 Semenyuta I, Kovalishyn V, Tanchuk V, Pilyo S, Zyabrev V, Blagodatnyy V, Trokhimenko O, Brovarets V, Metelytsia L. Comput. Biol. Chem. 2016; 65: 8
- 52 Kachaeva MV, Hodyna DM, Semenyuta IV, Pilyo SG, Prokopenko VM, Kovalishyn VV, Metelytsia LO, Brovarets VS. Comput. Biol. Chem. 2018; 74: 294
- 53 Xing C, Zhuang Y, Xu TH, Feng Z, Zhou XE, Chen M, Wang L, Meng X, Xue Y, Wang J, Liu H, McGuire TF, Zhao G, Melcher K, Zhang C, Xu HE, Xie XQ. Cell 2020; 180: 645
- 54 He L, Zhao Q, Qi J, Wang Y, Han W, Chen Z, Cong Y, Wang S. Proc. Natl. Acad. Sci. U. S. A. 2023; 120: e2209917120
- 55 Cai H, Guo S, Xu Y, Sun J, Li J, Xia Z, Jiang Y, Xie X, Xu HE. Nat. Commun. 2024; 15: 3252
- 56 Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature 2004; 428: 198
- 57 Wenglowsky S, Ren L, Ahrendt KA, Laird ER, Aliagas I, Alicke B, Buckmelter AJ, Choo EF, Dinkel V, Feng B, Gloor SL, Gould SE, Gross S, Gunzner-Toste J, Hansen JD, Hatzivassiliou G, Liu B, Malesky K, Mathieu S, Newhouse B, Raddatz NJ, Ran Y, Rana S, Randolph N, Risom T, Rudolph J, Savage S, Selby LT, Shrag M, Song K, Sturgis HL, Voegtli WC, Wen Z, Willis BS, Woessner RD, Wu W, Young WB, Grina J. ACS Med. Chem. Lett. 2011; 2: 342
- 58 Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schönbrunn E. ACS Chem. Biol. 2011; 6: 492
- 59 Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000; 28: 235
- 60 Trott O, Olson AJ. J. Comput. Chem. 2010; 31: 455
- 61 Marvin n. n. n. (5.2.4.), 201n (2009), ChemAxon (accessed Nov 21, 2025): http://www.chemaxon.com
- 62 Hanwell MD, Curtis DE, Lonie DC. J. Cheminform. 2012; 4: 4
- 63 Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009; 30: 2785
- 64 ADMETlab3 (accessed Nov 21, 2025): https://admetlab3.scbdd.com/server/evaluation
- 65 SwissADME (accessed Nov 21, 2025): http://www.swissadme.ch/index.php
- 66 Deep-PK deep learning for small molecule pharmacokinetic and toxicity prediction (accessed Nov 21, 2025): https://biosig.lab.uq.edu.au/deeppk/prediction
- 67 Introduction to MarvinView (accessed Nov 21, 2025): https://docs.chemaxon.com/display/lts-europium/introduction-to-marvinview.md
Corresponding Author
Publication History
Received: 01 September 2025
Accepted after revision: 07 November 2025
Accepted Manuscript online:
07 November 2025
Article published online:
01 December 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Kakkar S, Narasimhan B. BMC Chem. 2019; 13: 16
- 2 Joshi S, Mehra M, Singh R, Kakar S. Egypt. J. Basic Appl. Sci. 2023; 10: 218
- 3 Yan X, Jing W, Zhou L, Fan L, Wang X, Xu Z. Curr. Top. Med. Chem. 2020; 20: 1916
- 4 Sawant RS, Deventhiran P, Uppalwar SV, Sen AK. IJRPR 2024; 5: 1510
- 5 Kulkarni S, Kaur K, Jaitak V. Anti-cancer Agents Med. Chem. 2022; 22: 1859
- 6 El-Dydamony NM, Abdelnaby RM, Abdelhady R, Ali O, Fahmy MI, Eldeen R FakhrR, Helwa AA. J. Enzyme Inhib. Med. Chem. 2022; 37: 895
- 7 Reda N, Mohamed KO, Abdou K, Helwa AA, Elshewy A. Bioorg. Chem. 2024; 145: 107185
- 8 Al-Wahaibi LH, Abou-Zied HA, Hisham M, Beshr EA. M, Youssif BG. M, Bräse S, Hayallah AM, Abdel-Aziz M. Molecules 2023; 28: 6586
- 9 Saffari Z, Aryapour H, Akbarzadeh A, Foroumadi A, Jafari N, Farahnak Zarabi M, Farhangi A. Tumour Biol. 2014; 35: 5845
- 10 Ahmed NM, Mohamed MS, Awad SM, Abd El-Hameed RH, El-Tawab NA. A, Gaballah MS, Said AM. J. Enzyme Inhib. Med. Chem. 2024; 39: 2304625
- 11 Kachaeva MV, Pilyo SG, Demydchuk BA, Prokopenko VM, Zhirnov VV, Brovarets VS. Int. J. Curr. Res. 2018; 10: 69410
- 12 Kachaeva MV, Pilyo SG, Zhirnov VV, Brovarets VS. Med. Chem. Res. 2019; 28: 71
- 13 Severin O, Pilyo S, Kachaeva M, Zhirnov V, Hodyna D, Bahrieieva O, Brovarets V. ChemMedChem 2024; 19: e202300527
- 14 Severin O, Pilyo S, Semenyuta I, Kachaeva M, Zhirnov V, Brovarets V. Curr. Chem. Lett. 2025; 14: 159
- 15 Schole J, Schole C, Eikemeyer J. Z. Naturforsch., C 1994; 49: 657
- 16 Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Cells 2019; 8: 728
- 17 Kenny TC, Birsoy K. Cold Spring Harbor Perspect. Med. 2024; 14: a041534
- 18 Brusnakov M, Golovchenko O, Velihina Y, Liavynets O, Zhirnov V, Brovarets V. ChemMedChem 2022; 17: e202200319
- 19 Kondratyuk KM, Lukashuk EI, Golovchenko AV, Vasilenko AN, Brovarets VS. Russ. J. Gen. Chem. 2013; 83: 46
- 20 Pil’o SG, Brovarets VS, Vinogradova TK, Golovchenko AV, Drach BS. Russ. J. Gen. Chem. 2002; 72: 1714
- 21 Kachaeva MV, Hodyna M, Obernikhina NV, Pilyo SG, Kovalenko YS, Prokopenko VM, Kachkovsky OD, Brovarets VS. J. Heterocycl. Chem. 2019; 56: 3122
- 22 Obernikhina NV. OMCIJ 2019; 9: 555751
- 23a Lukovits I, Linert W. J. Chem. Inf. Comput. Sci. 1998; 38: 715
- 23b Chen L, Li T, Liu J, Shi Y, Wang H. PLoS One 2016; 11: e0167075
- 24 Whitley DC. J. Math. Chem. 1998; 23: 377
- 25 Agoni C, Olotu FA, Ramharack P, Soliman ME. J. Mol. Model. 2020; 26: 120
- 26 Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G, Casellas P. Eur. J. Biochem. 1993; 214: 173
- 27 Soto-Mercado V, Mendivil-Perez M, Jimenez-Del-Rio M, Fox JE, Velez-Pardo C. Leuk. Res. 2020; 95: 106389
- 28 Melén CM, Merrien M, Wasik AM, Panagiotidis G, Beck O, Sonnevi K, Junlén HR, Christensson B, Sander B, Wahlin BE. Leuk. Lymphoma 2022; 63: 1387
- 29 Zhang W, Zhang C, Liu F, Mao Y, Xu W, Fan T, Sun Q, He S, Chen Y, Guo W, Tan Y, Jiang Y. Sci. Rep. 2018; 8: 15753
- 30 Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. Immunol., Endocr. Metab. Agents Med. Chem. 2007; 7: 298
- 31 Peerzada MN, Dar MS, Verma S. Expert Opin. Ther. Pat. 2023; 33: 797
- 32 Maji L, Teli G, Raghavendra NM, Sengupta S, Pal R, Ghara A, Matada GS. P. Mol. Diversity 2024; 28: 2689
- 33 Marak BN, Dowarah J, Khiangte L, Singh VP. Eur. J. Med. Chem. 2020; 203: 112571
- 34 Lee K, Jang J, Seo S, Lim J, Kim WY. Chem. Sci. 2021; 13: 554
- 35 Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A, Chen X, Hou T, Cao D. Nucleic Acids Res. 2021; 49: W5
- 36 Patel D, Sethi N, Patel P, Shah S, Patel K. Eur. J. Pharm. Biopharm. 2024; 198: 114267
- 37 Gonçalves BM. F, Cardoso DS. P, Ferreira MJ. U. Molecules 2020; 25: 3364
- 38 Elfadadny A, El-Husseiny HM, Abugomaa A, Ragab RF, Mady EA, Aboubakr M, Samir H, Mandour AS, El-Mleeh A, El-Far AH, Abd El-AzizA. H, Elbadawy M. Environ. Sci. Pollut. Res. Int. 2021; 28: 49447
- 39 Husain A, Makadia V, Valicherla GR, Riyazuddin M, Gayen JR. Drug Dev. Res. 2022; 83: 825
- 40 Tian Y, Lei Y, Wang Y, Lai J, Wang J, Xia F. Int. J. Oncol. 2023; 63: 119
- 41 Guengerich FP. Annu. Rev. Pharmacol. Toxicol. 1999; 39: 1
- 42 Grever MR, Schepartz SA, Chabner BA. Semin. Oncol. 1992; 19: 622
- 43 Boyd MR, Paull KD. Drug Dev. Res. 1995; 34: 91
- 44 Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Jangley J, Cronisie P, Viagro-Wolff A, Gray-Goodrich M, Campell H, Boyd M. J. Natl. Cancer Inst. 1991; 83: 757
- 45 Data (accessed Nov 21, 2025): https://dtp.cancer.gov/discovery_development/nci-60/default.htm
- 46 Acton EM, Narayanan VL, Risbood PA, Shoemaker RH, Vistica DT, Boyd MR. J. Med. Chem. 1994; 37: 2185
- 47 Weerapreeyakul N, Nonpunya A, Barusrux S, Thitimetharoch T, Sripanidkulchai B. Chin. Med. 2012; 7: 15
- 48 History of the NCI-60 screen and COMPARE algorithm (accessed Nov 21, 2025): https://dtp.cancer.gov/databases_tools/compare.htm
- 49 Shoemaker RH. Nat. Rev. Cancer. 2006; 6: 813
- 50 Daina A, Michielin O, Zoete V. Nucleic Acids Res. 2019; 47: W357
- 51 Semenyuta I, Kovalishyn V, Tanchuk V, Pilyo S, Zyabrev V, Blagodatnyy V, Trokhimenko O, Brovarets V, Metelytsia L. Comput. Biol. Chem. 2016; 65: 8
- 52 Kachaeva MV, Hodyna DM, Semenyuta IV, Pilyo SG, Prokopenko VM, Kovalishyn VV, Metelytsia LO, Brovarets VS. Comput. Biol. Chem. 2018; 74: 294
- 53 Xing C, Zhuang Y, Xu TH, Feng Z, Zhou XE, Chen M, Wang L, Meng X, Xue Y, Wang J, Liu H, McGuire TF, Zhao G, Melcher K, Zhang C, Xu HE, Xie XQ. Cell 2020; 180: 645
- 54 He L, Zhao Q, Qi J, Wang Y, Han W, Chen Z, Cong Y, Wang S. Proc. Natl. Acad. Sci. U. S. A. 2023; 120: e2209917120
- 55 Cai H, Guo S, Xu Y, Sun J, Li J, Xia Z, Jiang Y, Xie X, Xu HE. Nat. Commun. 2024; 15: 3252
- 56 Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature 2004; 428: 198
- 57 Wenglowsky S, Ren L, Ahrendt KA, Laird ER, Aliagas I, Alicke B, Buckmelter AJ, Choo EF, Dinkel V, Feng B, Gloor SL, Gould SE, Gross S, Gunzner-Toste J, Hansen JD, Hatzivassiliou G, Liu B, Malesky K, Mathieu S, Newhouse B, Raddatz NJ, Ran Y, Rana S, Randolph N, Risom T, Rudolph J, Savage S, Selby LT, Shrag M, Song K, Sturgis HL, Voegtli WC, Wen Z, Willis BS, Woessner RD, Wu W, Young WB, Grina J. ACS Med. Chem. Lett. 2011; 2: 342
- 58 Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, Schönbrunn E. ACS Chem. Biol. 2011; 6: 492
- 59 Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000; 28: 235
- 60 Trott O, Olson AJ. J. Comput. Chem. 2010; 31: 455
- 61 Marvin n. n. n. (5.2.4.), 201n (2009), ChemAxon (accessed Nov 21, 2025): http://www.chemaxon.com
- 62 Hanwell MD, Curtis DE, Lonie DC. J. Cheminform. 2012; 4: 4
- 63 Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009; 30: 2785
- 64 ADMETlab3 (accessed Nov 21, 2025): https://admetlab3.scbdd.com/server/evaluation
- 65 SwissADME (accessed Nov 21, 2025): http://www.swissadme.ch/index.php
- 66 Deep-PK deep learning for small molecule pharmacokinetic and toxicity prediction (accessed Nov 21, 2025): https://biosig.lab.uq.edu.au/deeppk/prediction
- 67 Introduction to MarvinView (accessed Nov 21, 2025): https://docs.chemaxon.com/display/lts-europium/introduction-to-marvinview.md