
Subscribe to RSS
DOI: 10.1055/s-0042-1751411
A Cyclobutanol Ring-Expansion Approach to Oxygenated Carbazoles: Total Synthesis of Glycoborine, Carbazomycin A and Carbazomycin B
Abstract
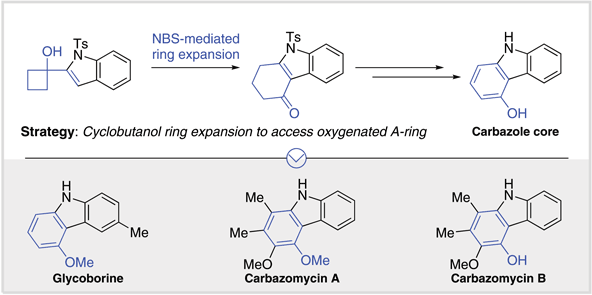
The transition-metal-free total syntheses of the oxygenated carbazole natural products glycoborine, carbazomycin A and carbazomycin B are reported. The key step involves an NBS-mediated cyclobutanol ring expansion to 4-tetralones for the preparation of the tricyclic carbazole core.
#
In the last few decades, the carbazole core has attracted the interest of synthetic organic chemists due to its versatile application in various fields of chemistry, such as the design of efficient photochemical materials or active pharmaceutical ingredients (APIs). The development of APIs, such as carazolol or carvedilol, is often inspired by initial hits from naturally occurring biologically active carbazoles (Scheme [1, a]). Interestingly, these marketed drugs contain oxygenated functionality at the 5-position, which is rarely found in naturally occurring carbazoles.
The first natural 5-oxygenated carbazole alkaloid, glycoborine, was reported by Chakravarty only two decades ago. It was isolated as a constituent in trace amounts (0.0001% w/w) of the petroleum ether extract of the roots of Glycosmis arborea (Scheme [1, a]),[1] and extracts of this plant have been used in traditional treatments of fever or liver diseases.[2] Given the low structural complexity of glycoborine, it has been a suitable target for synthetic chemists to test the applicability of their newly developed methodologies towards the carbazole core. In fact, the majority of these protocols focus on the assembly of the pyrrole core as the strategic step by using the Fischer indole synthesis,[1] [3] nickel-catalyzed reductive cyclization,[4] palladium-catalyzed cyclization of diarylamines,[5] or a variant of the Cadogan–Sundberg reaction (Scheme [1, b]).[6] [7] [8] [9] [10] [11] [12]
The carbazomycins are a family of naturally occurring carbazoles that also contain oxygenation on the same position (Scheme [1, a]). These compounds were isolated by Nakamura from the extract of cultured mycelia of Streptoverticillium ehimensis strain H1051-MY10,[13] [14] [15] and they are characterized by their unusual asymmetric substitution pattern, in which only one aromatic ring is fully substituted with electron-donating substituents. Several carbazomycins exhibit antifungal,[16] antibacterial[13] and anticancer activity;[17] [18] in addition, carbazomycin B displays inhibitory activity against 5-lipoxygenase (IC50 = 1.5 μg/mL)[19] and is active against malaria (IC50 = 2.37 μg/mL against Plasmodium falciparum, K1 multi-drug resistant strain).[20] Given these interesting biological effects, their total synthesis has received considerable attention (Scheme [1, b]).[21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] Elegant examples include an iron-mediated one-pot oxidative coupling by Knölker,[21] [22] [30] [31] a radical cyclization by Crich and Rumthao,[32] and most recently, a one-pot domino benzannulation between indole-3-ynones and nitroalkanes by Mehta.[34]
To complement these previously reported approaches to carbazole natural products, and encouraged by a recent total synthesis report utilizing a cyclobutanol ring expansion,[35] we were inspired to develop a strategy based on our previously developed cyclobutanol ring expansion methodology (Scheme [1, c]). In 2018, we reported that heteroaromatic-substituted cyclobutanols undergo regioselective ring expansion to 4-tetralones by treatment with N-bromosuccinimide in acetonitrile.[36] [37] [38] [39] [40] Following our group’s long-standing interest in the synthesis of biologically active natural products,[41–44] we now report the successful total syntheses of glycoborine, carbazomycin A and carbazomycin B.[45]
Our synthetic plan was guided by the oxygenation pattern of the carbazoles of interest (Scheme [1, c]). Thus, we hypothesized that the respective carbazole cores could be accessed by aromatization of a suitably substituted 4-tetralone intermediate 2. The key feature of our synthetic plan is the disconnection of the tricyclic intermediate to cyclobutanol 1, provided by the addition of protected indole to an appropriately substituted cyclobutanone.

With our synthetic plan laid out, we initiated our studies towards the total synthesis of glycoborine (Scheme [2]).[45] Synthesis of the core began with tosyl-protection of commercially available 5-methylindole (3) in 92% yield. Subsequent C2-lithiation with n-butyllithium, followed by addition to commercially available cyclobutanone (5) provided the cyclobutanol 6 in 88% yield on a decagram scale. This key intermediate was then converted into the 4-tetralone 7 by our previously reported cyclobutanol ring expansion in the presence of N-bromosuccinimide in acetonitrile. Notably, the 4-tetralone 7 was afforded on a multigram scale within 30 minutes.
To complete the total synthesis of glycoborine in short order, we next aimed to convert 4-tetralone 7 into the phenol 8 (see the Supporting Information). To our surprise, this proved to be a non-trivial quest: several aromatization conditions were examined, including bromination–elimination sequences, DDQ in toluene, or dichloromethane, and 10% Pd/C in boiling diphenyl ether. Yet, these conditions furnished the desired phenol in trace quantities only. Finally, we turned our attention to an iodine-mediated procedure reported by Tecle.[46] To our delight, treatment of 4-tetralone 7 with a superstoichiometric quantity of iodine in boiling methanol not only affected the desired aromatization but also afforded anisole 9 in 47% yield. Finally, removal of the N-tosyl protecting group with potassium hydroxide in boiling degassed ethanol delivered glycoborine in 85% yield.[47] Our synthetic sample exhibited identical spectroscopic properties to those of the natural isolate.[1]

We then turned our attention to the structurally more challenging carbazomycins A and B.[45] The synthetic sequence began with the preparation of trans-2,3-dimethylcyclobutanone (16) in 30% yield by a stereospecific thermal [2+2]-cycloaddition between trans-but-2-ene gas and the keteniminium salt of dimethylacetamide (15), followed by mild hydrolysis in boiling dichloromethane according to an adapted procedure by Ghosez.[48] [49] Although the isolated yield of the cyclobutanone was reduced by its volatility and that of trans-but-2-ene, we found the reaction to be scalable to gram-quantities nonetheless; producing sufficient quantities for the total synthesis (Scheme [3]). Next, addition of C2-lithiated Mbs-protected indole[50] to cyclobutanone 16 afforded key cyclobutanol 11 in 55% yield as a single undefined C1-epimer on a multi-gram scale. Treatment of cyclobutanol 11 with N-bromosuccinimide under our standard ring-expansion conditions afforded the trans-1,2-dimethylated 4-tetralone 12 as a single diastereomer in 50% yield within only five minutes.
In line with our previous studies, full stereochemical retention, as well as exclusive formation of the β,γ-substituted 4-tetralone (vs the α,β-substituted 4-tetralone) was observed (Scheme [3]). This regioselectivity is attributed to exclusive initial migration of the most-substituted carbon as the partial carbocation character in the transition state is stabilized by inductive effects and hyperconjugation.[51] [52] The migratory ability of carbon moieties in such migratory shifts has been well-studied and not only explains the observed regioselectivity, but also the retention of stereochemistry at the migrating center. A further 1,2-sigmatropic rearrangement—supported by the acylium-like character of the carbonyl group—affords the trans-1,2-dimethylated 4-tetralone after aromatization, intercepting Nishida’s route towards carbazomycins A and B in very short order (three vs eight synthetic steps in the longest linear sequence from indole).[28,53]
With the tricyclic backbone in hand, we turned our attention to installing the α-methoxy group. We initially followed Nishida’s described two-step protocol consisting of silyl enol ether formation by treatment of ketone 12 with Hünig’s base and trimethylsilyl trifluoromethylsulfonate in dichloromethane, followed by removal of volatiles and subjection to [bis(trifluoroacetoxy)iodo]benzene in methanol.[28] In our hands, this protocol returned starting material exclusively, which we reasoned to be a consequence of the conjugate acid of Hünig’s base removing the labile TMS group before the nucleophilic substitution with the hypervalent iodine species. To our delight, substitution of Hünig's base with lower boiling triethylamine, and inclusion of a mild aqueous work-up after silyl enol ether formation rendered the subsequent α-methoxylation successful to afford ketone 13 in 43% yield. Notably, the α-methoxylation proceeded stereospecifically to afford the cis-trans-substituted 4-tetralone exclusively (Scheme [3]).
Conversion into the natural products was then achieved following Nishida’s report (Scheme [3]).[28] Aromatization of ketone 13 with four equivalents of N-bromosuccinimide in THF afforded carbazole 14 in 44% yield. The importance of the Mbs-protecting group and the stereochemical relationship between the substituents on the tetralone ring for the success of the aromatization is key to note at this stage. Earlier studies within our group revealed that the N-tosyl-protected derivative failed to deliver the desired phenol under NBS-mediated conditions or alternative protocols, including Saegusa–Ito and iodine-mediated conditions. We attribute the aromatization of the Mbs-protected analogue to the greater electron-density induced by the alkoxy-resonance donor. Equally, we observed that, unlike the cis-trans-isomer, the cis-cis-isomer does not undergo aromatization under NBS-mediated conditions, confirming the necessity for control of the stereochemical relationship between the methyl groups from the outset of the synthetic sequence.
Finally, reduction of the sulfonyl protecting group using sodium anthracenide provided carbazomycin B in 57% yield, and this was converted, using Moody’s methylation conditions, into carbazomycin A in 87% yield (Scheme [3]).[26] [54] Our synthetic samples of carbazomycins A and B were spectroscopically identical to those of the natural isolates.[14]

In conclusion, we have reported a complementary strategy to oxygenated carbazoles. The total synthesis of glycoborine was achieved in a scalable, five-step and transition-metal-free sequence in 10% overall yield. Carbazomycin B was synthesized in six steps and converted into carbazomycin A in one further synthetic step. The reported syntheses are enabled by an NBS-mediated cyclobutanol ring expansion developed by our group, demonstrating its immediate applicability for medicinal chemistry or total synthesis applications. Extension to other classes of natural products, as well as biological evaluations are currently ongoing.
#
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We thank Pete Haycock (Imperial College London) and Dr. Lisa Haigh (Imperial College London) for NMR and mass spectrometric analysis, respectively.
Supporting Information
- Supporting information for this article is available online at https://doi.org/10.1055/s-0042-1751411.
- Supporting Information
-
References
- 1 Chakravarty AK, Sarkar T, Masuda K, Takey T, Doi H, Kotani E, Shiojima K. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2001; 40: 484
- 2 Chakravarty AK, Sarkar T, Masuda K, Shiojima K. Phytochemistry 1999; 50: 1263
- 3 Chakraborty A, Chowdhury BK, Jash SS, Biswas GK, Bhattacharyya SK, Bhattacharyya P. Phytochemistry 1992; 31: 2503
- 4 Yan Q, Gin E, Wasinska-Kalwa M, Banwell MG, Carr PD. J. Org. Chem. 2017; 82: 4148
- 5 Brütting C, Hesse R, Jäger A, Kataeva O, Schmidt AW, Knölker H.-J. Chem. Eur. J. 2016; 22: 16897
- 6 Gao H, Xu Q.-L, Yousufuddin M, Ess DH, Kürti L. Angew. Chem. Int. Ed. 2014; 53: 2701
- 7 Bhatthula BK. G, Kanchani JR, Arava VR, Subha MC. S. Tetrahedron 2019; 75: 874
- 8 Kuethe JT, Childers KG. Adv. Synth. Catal. 2008; 350: 1577
- 9 Nykaza TV, Ramirez A, Harrison TS, Luzung MR, Radosevich AT. J. Am. Chem. Soc. 2018; 140: 3103
- 10 Yang L, Zhang Y, Zou X, Lu H, Li G. Green Chem. 2018; 20: 1362
- 11 Yang L, Li H, Zhang H, Lu H. Eur. J. Org. Chem. 2016; 5611
- 12 Akiyama T, Wada Y, Yamada M, Shio Y, Honma T, Shimoda S, Tsuruta K, Tamenori Y, Haneoka H, Suzuki T, Harada K, Tsurugi H, Mashima K, Hasegawa J, Sato Y, Arisawa M. Org. Lett. 2020; 22: 7244
- 13 Sakano K.-I, Ishimaru K, Nakamura S. J. Antibiot. 1980; 33: 683
- 14 Sakano K.-I, Nakamura S. J. Antibiot. 1980; 33: 961
- 15 Nakamura S, Kaneda M, Sakano K, Kushi Y, Iitaka Y. Heterocycles 1981; 15: 993
- 16 Kaneda M, Naid T, Kitahara T, Nakamura S, Hirata T, Suga T. J. Antibiot. 1988; 41: 602
- 17 Nishiyama T, Hatae N, Yoshimura T, Takaki S, Abe T, Ishikura M, Hibino S, Choshi T. Eur. J. Med. Chem. 2016; 121: 561
- 18 Karwehl S, Jansen R, Huch V, Stadler M. J. Nat. Prod. 2016; 79: 369
- 19 Hook DJ, Yacobucci JJ, O’Connor S, Lee M, Kerns E, Krishnan B, Matson J, Hesler G. J. Antibiot. 1990; 43: 1347
- 20 Intaraudom C, Rachtawee P, Suvannakad R, Pittayakhajonwut P. Tetrahedron 2011; 67: 7593
- 21 Knölker H.-J, Bauermeister M. J. Chem. Soc., Chem. Commun. 1989; 1468
- 22 Knölker H.-J, Fröhner W. Tetrahedron Lett. 1999; 40: 6915
- 23 Markad SB, Argade NP. Org. Lett. 2014; 16: 5470
- 24 Schmidt AW, Reddy KR, Knölker H.-J. Chem. Rev. 2012; 112: 3193 ; and references cited therein
- 25 Knölker H.-J, Reddy KR. Chem. Rev. 2002; 102: 4303 ; and references cited therein
- 26 Moody CJ, Shah P. J. Chem. Soc., Perkin Trans. 1 1989; 376
- 27 Beccalli EM, Marchesini A, Pilati T. Tetrahedron 1996; 52: 3029
- 28 Wu S, Harada S, Morikawa T, Nishida A. Chem. Pharm. Bull. 2018; 66: 178
- 29 Hibino S, Tonari A, Choshi T, Sugino E. Heterocycles 1993; 35: 441
- 30 Knölker H.-J, Bauermeister M, Bläser D, Boese R, Pannek J.-B. Angew. Chem. 1989; 101: 225
- 31 Knölker H.-J, Bauermeister M. Helv. Chim. Acta 1993; 76: 2500
- 32 Crich D, Rumthao S. Tetrahedron 2004; 60: 1513
- 33 Clive DL. J, Etkin N, Joseph T, Lown JW. J. Org. Chem. 1993; 58: 2442
- 34 Singh S, Samineni R, Pabbaraja S, Mehta G. Org. Lett. 2019; 21: 3372
- 35 Leclair A, Wang Q, Zhu J. ACS Catal. 2022; 12: 1209
- 36 Natho P, Kapun M, Allen LA. T, Parsons PJ. Org. Lett. 2018; 20: 8030
- 37 Natho P, Allen LA. T, White AJ. P, Parsons PJ. J. Org. Chem. 2019; 84: 9611
- 38 Natho P, Rouse AB, Greenfield JL, Allen LA. T, White AJ. P, Yang Z, Parsons PJ. Tetrahedron 2020; 76: 131636
- 39 Natho P, Allen LA. T, Parsons PJ. Tetrahedron Lett. 2020; 61: 151695
- 40 Petti A, Natho P, Lam K, Parsons PJ. Eur. J. Org. Chem. 2021; 854
- 41 Parsons PJ, Jones DR, Walsh LJ, Allen LA. T, Onwubiko A, Preece L, Board J, White AJ. P. Org. Lett. 2017; 19: 2533
- 42 Chandler M, Parsons PJ. J. Chem. Soc., Chem. Commun. 1984; 322
- 43 Parsons PJ, Pennicott L, Eshelby J, Goessman M, Highton A, Hitchcock P. J. Org. Chem. 2007; 72: 9387
- 44 Greenwood E, Hitchcock P, Parsons P. Tetrahedron 2003; 59: 3307
- 45 Natho P. The Discovery and Application of Metal-Free Cyclobutanol Ring Expansion Reactions, Ph.D. Thesis; Imperial College London, UK, 2021.
- 46 Tecle H, Bergmeier SC, Wise LD, Hershenson FM, Coughenour LL, Heffner TG. J. Heterocycl. Chem. 1989; 26: 1125
- 47 5-Methoxy-3-methyl-9H-carbazole (Glycoborine) To a solution of 5-methoxy-3-methyl-9-tosyl-9H-carbazole (9) (20 mg, 0.055 mmol) in degassed ethanol (5 mL) was added finely ground potassium hydroxide (15 mg, 0.27 mmol) in one portion. The resulting solution was heated at reflux for 20 hours, before cooling to room temperature and removal of the volatiles under reduced pressure. The concentrate was dissolved in ethyl acetate (10 mL), and the organic layer was washed with deionized water (2 × 10 mL) and saturated aqueous sodium chloride solution (15 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to afford the title compound (10 mg, 0.047 mmol, 85%) as a white solid (mp 135.1–136.8 °C [Lit.1 155–156 °C; PE–CHCl3]). IR (neat): 3405, 3042, 3004, 2948, 2915, 2837, 1606, 1586, 1506, 1459, 1260, 1100 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.14 (br s, 1 H), 7.91 (br s, 1 H), 7.33 (t, J = 8.0 Hz, 1 H), 7.28 (dd, J = 8.2, 0.7 Hz, 1 H), 7.22 (ddd, J = 8.2, 1.7, 0.7 Hz, 1 H), 7.01 (dd, J = 8.2, 0.7 Hz, 1 H), 6.67 (dd, J = 8.0, 0.7 Hz, 1 H), 4.09 (s, 3 H), 2.55 (d, J = 0.8 Hz, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 156.2, 141.2, 136.9, 128.9, 126.5, 126.2, 123.0, 122.8, 112.5, 109.6, 103.5, 100.2, 55.4, 21.5. HRMS (APCI): m/z [M + H]+ calcd for C14H14ON: 212.1070; found: 212.1067. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.1
- 48 Falmagne J.-B, Escudero J, Taleb-Sahraoui S, Ghosez L. Angew. Chem. 1981; 93: 926
- 49 Houge C, Frisque-Hesbain AM, Mockel A, Ghosez L, Declercq JP, Germain G, Van Meerssche M. J. Am. Chem. Soc. 1982; 104: 2920
- 50 Ramalingan C, Lee I.-S, Kwak Y.-W. Chem. Pharm. Bull. 2009; 57: 591
- 51 Nagakura I, Savary DN.-H, Schlosser M. Helv. Chim. Acta 1980; 63: 1257
- 52 ten Brink G.-J, Arends IW. C. E, Sheldon RA. Chem. Rev. 2004; 104: 4105
- 53 Wu S, Harada S, Morikawa T, Nishida A. Tetrahedron: Asymmetry 2017; 28: 1083
- 54 Carbazomycin B Carbazomycin B was synthesized according to a procedure by Nishida and co-workers.28 A solution of anthracene (65 mg, 0.36 mmol) in THF (1.5 mL) was purged with nitrogen for 10 minutes, before sodium metal (13 mg, 0.57 mmol) was added. The suspension was stirred at room temperature for 30 minutes, before being sonicated for a further 20 minutes. The dark blue solution was removed from the sonication bath, and a solution of 3-methoxy-9-((4-methoxyphenyl)sulfonyl)-1,2-dimethyl-9H-carbazol-4-ol (14) (30 mg, 0.073 mmol) in THF (1 mL) was added in one portion. The resulting green solution was stirred at room temperature for 1 hour, before it was diluted with dichloromethane (10 mL), and deionized water (10 mL) was added. The aqueous layer was separated and extracted with dichloromethane (3 × 10 mL). The combined organic extracts were washed with deionized water (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (10–30% EtOAc/pentane) to afford the title compound (10 mg, 0.041 mmol, 57%) as a white solid. IR (neat): 3425, 3053, 2988, 2923, 2854, 1638, 1612, 1500, 1453, 1411, 1321, 1300, 1144, 1083, 1003 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.25 (m, 1 H), 7.78 (br s, 1 H), 7.43–7.32 (m, 2 H), 7.22 (ddd, J = 8.1, 6.8, 1.4 Hz, 1 H), 6.06 (s, 1 H), 3.83 (s, 3 H), 2.40 (s, 3 H), 2.37 (s, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 142.0, 139.2, 138.4, 136.7, 127.0, 124.8, 123.3, 122.6, 119.5, 110.0, 109.3, 109.3, 61.5, 13.2, 12.8. HRMS (APCI): m/z [M + H]+ calcd for C15H16NO2: 242.1176; found: 242.1182. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.28 Carbazomycin A Carbazomycin A was synthesized according to a procedure by Moody and Shah.26 To a solution of carbazomycin B (8.0 mg, 0.033 mmol) in acetone (2 mL) was added dried potassium carbonate (50 mg, 0.36 mmol) and iodomethane (0.3 mL, 4.8 mmol) sequentially. The resulting reaction mixture was then heated at reflux for 3 hours, before it was allowed to cool to room temperature, and diluted with dichloromethane (5 mL). The organic phase was separated and washed with deionized water (3 × 5 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (10% EtOAc/pentane) to afford the title compound (7.3 mg, 0.029 mmol, 87%) as a yellow oil. IR (neat): 3436, 3351, 2998, 2989, 2930, 1610, 1498, 1455, 1395, 1293, 1088, 1051 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.23 (m, 1 H), 7.83 (s, 1 H), 7.45–7.33 (m, 1 H), 7.22 (ddd, J = 8.0, 6.8, 1.5 Hz, 1 H), 4.11 (s, 1 H), 3.90 (s, 1 H), 2.41 (s, 1 H), 2.39 (s, 1 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 146.0, 144.5, 139.4, 136.4, 128.8, 125.1, 122.9, 122.5, 119.5, 114.4, 113.5, 110.3, 61.1, 60.6, 13.7, 12.6. IR (neat): 3436, 3351, 2998, 2989, 2930, 1610, 1498, 1455, 1395, 1293, 1088, 1051 cm–1. HRMS (APCI): m/z [M + H]+ calcd for C16H18NO2: 256.1332; found: 256.1329. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.28
Corresponding Author
Publication History
Received: 27 November 2022
Accepted after revision: 28 December 2022
Article published online:
24 January 2023
© 2023. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Chakravarty AK, Sarkar T, Masuda K, Takey T, Doi H, Kotani E, Shiojima K. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 2001; 40: 484
- 2 Chakravarty AK, Sarkar T, Masuda K, Shiojima K. Phytochemistry 1999; 50: 1263
- 3 Chakraborty A, Chowdhury BK, Jash SS, Biswas GK, Bhattacharyya SK, Bhattacharyya P. Phytochemistry 1992; 31: 2503
- 4 Yan Q, Gin E, Wasinska-Kalwa M, Banwell MG, Carr PD. J. Org. Chem. 2017; 82: 4148
- 5 Brütting C, Hesse R, Jäger A, Kataeva O, Schmidt AW, Knölker H.-J. Chem. Eur. J. 2016; 22: 16897
- 6 Gao H, Xu Q.-L, Yousufuddin M, Ess DH, Kürti L. Angew. Chem. Int. Ed. 2014; 53: 2701
- 7 Bhatthula BK. G, Kanchani JR, Arava VR, Subha MC. S. Tetrahedron 2019; 75: 874
- 8 Kuethe JT, Childers KG. Adv. Synth. Catal. 2008; 350: 1577
- 9 Nykaza TV, Ramirez A, Harrison TS, Luzung MR, Radosevich AT. J. Am. Chem. Soc. 2018; 140: 3103
- 10 Yang L, Zhang Y, Zou X, Lu H, Li G. Green Chem. 2018; 20: 1362
- 11 Yang L, Li H, Zhang H, Lu H. Eur. J. Org. Chem. 2016; 5611
- 12 Akiyama T, Wada Y, Yamada M, Shio Y, Honma T, Shimoda S, Tsuruta K, Tamenori Y, Haneoka H, Suzuki T, Harada K, Tsurugi H, Mashima K, Hasegawa J, Sato Y, Arisawa M. Org. Lett. 2020; 22: 7244
- 13 Sakano K.-I, Ishimaru K, Nakamura S. J. Antibiot. 1980; 33: 683
- 14 Sakano K.-I, Nakamura S. J. Antibiot. 1980; 33: 961
- 15 Nakamura S, Kaneda M, Sakano K, Kushi Y, Iitaka Y. Heterocycles 1981; 15: 993
- 16 Kaneda M, Naid T, Kitahara T, Nakamura S, Hirata T, Suga T. J. Antibiot. 1988; 41: 602
- 17 Nishiyama T, Hatae N, Yoshimura T, Takaki S, Abe T, Ishikura M, Hibino S, Choshi T. Eur. J. Med. Chem. 2016; 121: 561
- 18 Karwehl S, Jansen R, Huch V, Stadler M. J. Nat. Prod. 2016; 79: 369
- 19 Hook DJ, Yacobucci JJ, O’Connor S, Lee M, Kerns E, Krishnan B, Matson J, Hesler G. J. Antibiot. 1990; 43: 1347
- 20 Intaraudom C, Rachtawee P, Suvannakad R, Pittayakhajonwut P. Tetrahedron 2011; 67: 7593
- 21 Knölker H.-J, Bauermeister M. J. Chem. Soc., Chem. Commun. 1989; 1468
- 22 Knölker H.-J, Fröhner W. Tetrahedron Lett. 1999; 40: 6915
- 23 Markad SB, Argade NP. Org. Lett. 2014; 16: 5470
- 24 Schmidt AW, Reddy KR, Knölker H.-J. Chem. Rev. 2012; 112: 3193 ; and references cited therein
- 25 Knölker H.-J, Reddy KR. Chem. Rev. 2002; 102: 4303 ; and references cited therein
- 26 Moody CJ, Shah P. J. Chem. Soc., Perkin Trans. 1 1989; 376
- 27 Beccalli EM, Marchesini A, Pilati T. Tetrahedron 1996; 52: 3029
- 28 Wu S, Harada S, Morikawa T, Nishida A. Chem. Pharm. Bull. 2018; 66: 178
- 29 Hibino S, Tonari A, Choshi T, Sugino E. Heterocycles 1993; 35: 441
- 30 Knölker H.-J, Bauermeister M, Bläser D, Boese R, Pannek J.-B. Angew. Chem. 1989; 101: 225
- 31 Knölker H.-J, Bauermeister M. Helv. Chim. Acta 1993; 76: 2500
- 32 Crich D, Rumthao S. Tetrahedron 2004; 60: 1513
- 33 Clive DL. J, Etkin N, Joseph T, Lown JW. J. Org. Chem. 1993; 58: 2442
- 34 Singh S, Samineni R, Pabbaraja S, Mehta G. Org. Lett. 2019; 21: 3372
- 35 Leclair A, Wang Q, Zhu J. ACS Catal. 2022; 12: 1209
- 36 Natho P, Kapun M, Allen LA. T, Parsons PJ. Org. Lett. 2018; 20: 8030
- 37 Natho P, Allen LA. T, White AJ. P, Parsons PJ. J. Org. Chem. 2019; 84: 9611
- 38 Natho P, Rouse AB, Greenfield JL, Allen LA. T, White AJ. P, Yang Z, Parsons PJ. Tetrahedron 2020; 76: 131636
- 39 Natho P, Allen LA. T, Parsons PJ. Tetrahedron Lett. 2020; 61: 151695
- 40 Petti A, Natho P, Lam K, Parsons PJ. Eur. J. Org. Chem. 2021; 854
- 41 Parsons PJ, Jones DR, Walsh LJ, Allen LA. T, Onwubiko A, Preece L, Board J, White AJ. P. Org. Lett. 2017; 19: 2533
- 42 Chandler M, Parsons PJ. J. Chem. Soc., Chem. Commun. 1984; 322
- 43 Parsons PJ, Pennicott L, Eshelby J, Goessman M, Highton A, Hitchcock P. J. Org. Chem. 2007; 72: 9387
- 44 Greenwood E, Hitchcock P, Parsons P. Tetrahedron 2003; 59: 3307
- 45 Natho P. The Discovery and Application of Metal-Free Cyclobutanol Ring Expansion Reactions, Ph.D. Thesis; Imperial College London, UK, 2021.
- 46 Tecle H, Bergmeier SC, Wise LD, Hershenson FM, Coughenour LL, Heffner TG. J. Heterocycl. Chem. 1989; 26: 1125
- 47 5-Methoxy-3-methyl-9H-carbazole (Glycoborine) To a solution of 5-methoxy-3-methyl-9-tosyl-9H-carbazole (9) (20 mg, 0.055 mmol) in degassed ethanol (5 mL) was added finely ground potassium hydroxide (15 mg, 0.27 mmol) in one portion. The resulting solution was heated at reflux for 20 hours, before cooling to room temperature and removal of the volatiles under reduced pressure. The concentrate was dissolved in ethyl acetate (10 mL), and the organic layer was washed with deionized water (2 × 10 mL) and saturated aqueous sodium chloride solution (15 mL), dried over magnesium sulfate, filtered, and concentrated under reduced pressure to afford the title compound (10 mg, 0.047 mmol, 85%) as a white solid (mp 135.1–136.8 °C [Lit.1 155–156 °C; PE–CHCl3]). IR (neat): 3405, 3042, 3004, 2948, 2915, 2837, 1606, 1586, 1506, 1459, 1260, 1100 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.14 (br s, 1 H), 7.91 (br s, 1 H), 7.33 (t, J = 8.0 Hz, 1 H), 7.28 (dd, J = 8.2, 0.7 Hz, 1 H), 7.22 (ddd, J = 8.2, 1.7, 0.7 Hz, 1 H), 7.01 (dd, J = 8.2, 0.7 Hz, 1 H), 6.67 (dd, J = 8.0, 0.7 Hz, 1 H), 4.09 (s, 3 H), 2.55 (d, J = 0.8 Hz, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 156.2, 141.2, 136.9, 128.9, 126.5, 126.2, 123.0, 122.8, 112.5, 109.6, 103.5, 100.2, 55.4, 21.5. HRMS (APCI): m/z [M + H]+ calcd for C14H14ON: 212.1070; found: 212.1067. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.1
- 48 Falmagne J.-B, Escudero J, Taleb-Sahraoui S, Ghosez L. Angew. Chem. 1981; 93: 926
- 49 Houge C, Frisque-Hesbain AM, Mockel A, Ghosez L, Declercq JP, Germain G, Van Meerssche M. J. Am. Chem. Soc. 1982; 104: 2920
- 50 Ramalingan C, Lee I.-S, Kwak Y.-W. Chem. Pharm. Bull. 2009; 57: 591
- 51 Nagakura I, Savary DN.-H, Schlosser M. Helv. Chim. Acta 1980; 63: 1257
- 52 ten Brink G.-J, Arends IW. C. E, Sheldon RA. Chem. Rev. 2004; 104: 4105
- 53 Wu S, Harada S, Morikawa T, Nishida A. Tetrahedron: Asymmetry 2017; 28: 1083
- 54 Carbazomycin B Carbazomycin B was synthesized according to a procedure by Nishida and co-workers.28 A solution of anthracene (65 mg, 0.36 mmol) in THF (1.5 mL) was purged with nitrogen for 10 minutes, before sodium metal (13 mg, 0.57 mmol) was added. The suspension was stirred at room temperature for 30 minutes, before being sonicated for a further 20 minutes. The dark blue solution was removed from the sonication bath, and a solution of 3-methoxy-9-((4-methoxyphenyl)sulfonyl)-1,2-dimethyl-9H-carbazol-4-ol (14) (30 mg, 0.073 mmol) in THF (1 mL) was added in one portion. The resulting green solution was stirred at room temperature for 1 hour, before it was diluted with dichloromethane (10 mL), and deionized water (10 mL) was added. The aqueous layer was separated and extracted with dichloromethane (3 × 10 mL). The combined organic extracts were washed with deionized water (20 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (10–30% EtOAc/pentane) to afford the title compound (10 mg, 0.041 mmol, 57%) as a white solid. IR (neat): 3425, 3053, 2988, 2923, 2854, 1638, 1612, 1500, 1453, 1411, 1321, 1300, 1144, 1083, 1003 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.25 (m, 1 H), 7.78 (br s, 1 H), 7.43–7.32 (m, 2 H), 7.22 (ddd, J = 8.1, 6.8, 1.4 Hz, 1 H), 6.06 (s, 1 H), 3.83 (s, 3 H), 2.40 (s, 3 H), 2.37 (s, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 142.0, 139.2, 138.4, 136.7, 127.0, 124.8, 123.3, 122.6, 119.5, 110.0, 109.3, 109.3, 61.5, 13.2, 12.8. HRMS (APCI): m/z [M + H]+ calcd for C15H16NO2: 242.1176; found: 242.1182. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.28 Carbazomycin A Carbazomycin A was synthesized according to a procedure by Moody and Shah.26 To a solution of carbazomycin B (8.0 mg, 0.033 mmol) in acetone (2 mL) was added dried potassium carbonate (50 mg, 0.36 mmol) and iodomethane (0.3 mL, 4.8 mmol) sequentially. The resulting reaction mixture was then heated at reflux for 3 hours, before it was allowed to cool to room temperature, and diluted with dichloromethane (5 mL). The organic phase was separated and washed with deionized water (3 × 5 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified by flash column chromatography (10% EtOAc/pentane) to afford the title compound (7.3 mg, 0.029 mmol, 87%) as a yellow oil. IR (neat): 3436, 3351, 2998, 2989, 2930, 1610, 1498, 1455, 1395, 1293, 1088, 1051 cm–1. 1H NMR (400 MHz, CDCl3): δ = 8.23 (m, 1 H), 7.83 (s, 1 H), 7.45–7.33 (m, 1 H), 7.22 (ddd, J = 8.0, 6.8, 1.5 Hz, 1 H), 4.11 (s, 1 H), 3.90 (s, 1 H), 2.41 (s, 1 H), 2.39 (s, 1 H). 13C{1H} NMR (101 MHz, CDCl3): δ = 146.0, 144.5, 139.4, 136.4, 128.8, 125.1, 122.9, 122.5, 119.5, 114.4, 113.5, 110.3, 61.1, 60.6, 13.7, 12.6. IR (neat): 3436, 3351, 2998, 2989, 2930, 1610, 1498, 1455, 1395, 1293, 1088, 1051 cm–1. HRMS (APCI): m/z [M + H]+ calcd for C16H18NO2: 256.1332; found: 256.1329. The spectroscopic data (1H NMR, 13C{1H} NMR and IR) are consistent with the literature.28