RSS-Feed abonnieren
DOI: 10.1055/a-1987-6464
Versatile Utility of Cp*Co(III) Catalysts in C–H Amination under Inner- and Outer-Sphere Pathway

Dedicated to Professor Masahiro Murakami
Abstract
This Account describes the recent advances in our research program toward the development of cobalt-catalyzed C–H amidation reactions. In particular, synthetic versatilities of obtainable amino products shown to be achieved on the basis of two distinctive mechanistic scaffolds; inner- and outer-sphere pathways. It highlights our approaches to transit the modes of C–N bond formation by introduction of bidentate LX-type ligands into Cp*Co(III) precursors, thereby broadly expanding the scope of amination reactions.
1 Introduction
2 Cp*Co-Catalyzed Inner-Sphere C–H Amidation
3 Cp*Co-Catalyzed Outer-Sphere C–H Amidation
3.1 C(sp2)–N Bond Formation
3.2 C(sp3)–N Bond Formation
4 Conclusion
#
Key words
cobalt catalysis - C–H amidation - chelation-assisted amidation - nitrenoid insertion - site-selective amidationBiographical Sketches

Sukbok Chang is a Director at IBS and a professor at KAIST. In 1996, he earned his PhD in organic chemistry at Harvard University under the supervision of Professor Eric N. Jacobsen. After postdoctoral experience with Professor Robert H. Grubbs at Caltech, he joined Ewha Womans University in Seoul, Korea as an assistant professor in 1998 and then moved to KAIST in 2002. Since 2012 he has been the director of the Center for Catalytic Hydrocarbon Functionalizations at IBS. His research interests are on the development, understanding, and synthetic applications of transition-metal catalysis.

Jeonghyo Lee received his PhD in medicinal chemistry at the University of Michigan under the supervision of Professor Pavel Nagorny in 2019. He then joined Professor Sukbok Chang’s research group at the Institute for Basic Science (IBS) as a postdoctoral research fellow. His main research interest is the utilization of transition metals in the development of organic synthetic methodologies.
Introduction
C–H functionalization has emerged as one of the most efficient and straightforward approaches to construct carbon–heteroatom bonds directly from C–H bonds.[1] This strategy is advantageous especially from an atom economy and sustainability point of view as it does not require pre-functionalization of the substrates. Although it may offer more direct access to target functionality, selective activation of potentially reactive C–H bonds has remained a great challenge.[2] Along with great advances in organometallic chemistry, the modern application of transition-metal complexes in the catalytic C–H functionalizations has ushered in a new era in synthetic chemistry by providing precision to control selectivity and high reaction efficiency in a wide variety of chemical transformations.[3]
Among a range of transition-metal-based catalytic systems, Cp*M(III) complexes (Cp* = pentamethyl-cyclopentadienyl) centered on iridium and rhodium, in particular, have found pervasive utilities as an effective catalyst for various C–H functionalization reactions.[4] However, the environmental and economic concerns associated with the use of expensive and nongreen precious metals prompted the search for milder alternatives. In this context, the pioneering work of Matsunaga and Kanai on the use of [Cp*Co(III)] catalyst for the direct C(sp2)–H alkylation (Scheme [1]) provided a promising direction for sustainable and efficient catalysis.[5]

The Cp*Co(III) system is not only favorable from an economical or sustainable standpoint, but the unique properties of the cobalt metal core also lead to better efficiencies for certain types of functionalization reactions when even compared to the Ir or Rh counterparts.[6] For example, Glorius and co-workers reported a comparative study on the catalytic activity of group 9 Cp*M(III) toward the C–H amidation and cyclization cascade of aryl imidates, whereby the efficiency of Cp*Co had been proven superior in comparison to the Ir or Rh congeners (Scheme [2a]).[7] The authors attributed such an observation to the relatively stronger Lewis acidity of cobalt that may facilitate the cyclization reactivity, as well as to its smaller ionic radius that allows for the circumvention of the unwanted over-amidation pathway presumably via steric perturbations.

In addition, the inherent physicochemical property of cobalt metal arising from the d-electronic structure gave rise to new reactivity not seen in conventional catalytic reactions. Using a cobalt-porphyrin catalytic system, the Zhang group highlighted the involvement of cobalt-based metalloradical catalysis for the direct amination of electron-poor C–H bonds, which are difficult to achieve with the widely studied Rh2 or other closed-shell systems (Scheme [2b]).[8]
In 2014, the versatile application of the Cp*Co(III) catalytic system in various C–H functionalizations was recognized by Glorius and colleagues through their research work.[9] In this study, C–H cyanation, halogenation, and allylation reactions were realized for the first time by Cp*Co(III)-catalyzed formal SN-type reactions (Scheme [3]). This catalytic procedure was successfully applied to various types of sp2 carbons, including arenes, heteroarenes, and alkenes, thereby resulting in value-added organo nitriles, halides, as well as allylated indoles.

The cobalt(III) metal with Cp-type ligands has also served as a productive platform for the catalytic enantioselective C–H functionalization reactions.[10] The first successful application of Cp*Co(III) complexes for asymmetric C–H functionalization was proposed by Ackermann in 2018 (Scheme [4a]). In this work, the cooperative catalytic systems between Cp*Co(III) and chiral carboxylic acid were employed to promote the enantioselective C–H alkylation on indoles in a high position- and regioselective manner.[11] Cramer and co-workers revealed that the Cp* variant of the cobalt(III) complex also enabled asymmetric C–H functionalization (Scheme [4b]).[12] The Co(III) complex equipped with a trisubstituted chiral cyclopentadienyl ligand was found to be effective to provide high enantioselectivity as well as regioselectivity in the synthesis of dihydroisoquinolones from N-chlorobenzamides and alkenes, outperforming the best rhodium(III)-based methods for this type of reaction.

As a consequence of the synthetic advantages and distinctive activity, the Cp*Co catalytic systems have exploded in use for a wide range of other C–H functionalization reactions[13] as well, which have been comprehensively summarized in recent review articles by Yu,[14] Ackermann,[15] and Matsunaga.[16] Our group joined the historical evolution of this research area at a relatively early stage, particularly in the field of C–H amidation. In this Account, we present our own efforts on the recent development of Cp*Co-catalyzed C–N bond formation, which are discussed in two separate sections based on the two distinct mechanistic pathways: inner- and outer-sphere C–H functionalization (Scheme [5]).

The inner-sphere mechanism, also denoted as ‘C–H activation catalysis’ (depicted in red), is postulated to take place via C–H metalation to initially construct a cobaltacycle II. The corresponding cobalt complex in turn oxidatively activates an amino source to result in a cobalt-nitrenoid III, which then inserts into the internal Co–C bond to forge a new C–N bond. In this reaction pathway, the cobalt metal interconnects the key C–H activation and C–N bond-forming processes, so is referred as the inner-sphere pathway. In the outer-sphere pathway (illustrated in blue), in sharp contrast, an initially generated cobalt-nitrenoid species V directly interacts with the C–H bonds of substrate for the C–N bond formation. This mechanistic manifold involves ‘C–H insertion catalysis’ in that the C–H bond functionalization occurs at the nitrenoid moiety.
# 2
Cp*Co-Catalyzed Inner-Sphere C–H Amidation
In 2014, the viability of Cp*Co catalyst for C–H amination was disclosed for the first time by Matsunaga and Kanai (Scheme [6]).[17] They demonstrated that a readily available cobalt complex, Cp*Co(CO)I2, was efficient for the C2-selective directed C–H amidation of indoles using sulfonyl azides as an amino source. Despite the novelty and high efficiency of this transformation, the limited substrate scope and harsh reaction conditions (high temperatures, 100 °C) implied that there would be room for improvement in Co-catalyzed C–H amination. In this regard, we became interested in devising competent amino sources that can confer direct C–N bond formation from various C–H bonds under mild Cp*Co catalytic conditions.


In 2015, our group reported the utilization of O-acylcarbamates as a convenient amidating source in a Cp*Co(III)-catalyzed C–H amidation of arenes via chelation-assisted regiocontrol (Scheme [7]).[18] Under mild and external oxidant-free conditions, Co(III) catalysts exhibited high amidation efficiency for the construction of synthetically versatile N-aryl carbamate products, with excellent compatibility for a wide range of arenes possessing various substituents and directing groups such as pyridine, isoquinoline, pyrimidine, and purines.
A catalytic cycle of this amidation reaction was proposed to start via the generation of a cationic cobalt species VI, which can facilitate the C–H bond activation to form a 5-membered cobaltacycle VII (Scheme [8]). Coordination of an amidating reagent to the metallacycle followed by oxidative activation and insertion of the resulting nitrene species IX into the arene gives a Co(III)-amido species X. Subsequent protonolysis of X provides the final product with the regeneration of the active cobalt species VI.

The success in the Co-catalyzed inner-sphere C–H amidation prompted us to further expand the Cp*Co system using alternative nitrogen sources. When dioxazolones[19] were used as an amino reagent, low catalyst loading (1 mol%) of Cp*Co(III) was sufficient for the effective C–H amidation of anilides, relatively challenging arene substrates that were previously absent in the Cp*Co-catalyzed C–H functionalization (Scheme [9]).[20] A comparative study on the catalytic activity with Cp*Rh(III) and Cp*Ir(III) complexes disclosed that the efficiency of the cobalt system was superior in the desired C–H amidation of anilides when compared to that of the other group 9 analogues. The cobalt catalyst system could also be readily applied to a wide range of alternative substrate classes, including benzamides, phenylpyridines, phenyl pyrazoles, thiophenylpyridines, and benzoquinonlines to provide the desired amide products (Scheme [10]).


Utilizing robust dioxazolones as an amidating agent, Dixon, Seayad, and co-workers demonstrated the first example of Cp*Co(III)-catalyzed C(sp3)–H amidation in the presence of a thioamide as a directing group (Scheme [11a]).[21] This method was later further expanded to an asymmetric transformation using a hybrid catalytic system consisted of achiral cyclopentadienyl and chiral carboxylic acid by Yoshino and Matsunaga (Scheme [11b]).[22]
# 3
Cp*Co-Catalyzed Outer-Sphere C–H Amidation
Since the seminal work of Breslow in 1982 on the catalytic transfer of nitrene species for the amidation of cyclohexane,[23] transition-metal-catalyzed group transfer of metal-bound nitrene has emerged as one of the most powerful and efficient strategies for the construction of carbon–nitrogen bonds in organic synthesis.[24] In 2018, diverting our efforts from the inner-sphere C–H amidation reactions[25] to achieve an alternative strategy with group 9 Cp*M complexes, we established a novel outer-sphere C–H amidation process leveraging the intermediacy of Ir-acylnitrenoid species (Scheme [12]).[26] The use of Cp*Ir(III) bearing a strong σ-donating bidentate (L,X-type) ligand allowed for the desired intramolecular C–H amidation with a dioxazolone motif as the nitrene precursor, thereby leading to the efficient synthesis of γ-lactams while precluding the unwanted Curtius-type rearrangement pathway. We rationalized that the electron-donating ligand plays a pivotal role in effectively suppressing the Curtius decomposition pathway as it is closely related to the elevation of energy barrier of Curtius rearrangement, which is sensitive to changes in the partial charge of the metal center.


This milestone outer-sphere strategy has spurred the development of group-transfer protocols based on the piano-stool metal-nitrenoid intermediates for a wide range of C–H amidation reactions.[27] For instance, this mechanistic platform has proven successful for the chiral γ‑lactam synthesis,[28] unconventional C(sp 2)–N bond formation via spirocyclization,[29] oxyamidation of olefin,[30] haloamidation of alkynes,[31] migratory amidation of alkenyl alcohols,[32] chemodivergent C–H amidation,[33] benzylic selective C–H amidation reactions,[34] and selective α-amidation of esters.[35] Driven by the success of outer-sphere amidation approaches using iridium or ruthenium, we envisioned cobalt could also be implemented in the piano-stool catalytic system for effective group transfer of the putative nitrenoid intermediacy.
The Cp*Co(III)(κ2-N,O chelate) complexes turned out to be readily accessible by mixing a solution of dimeric precursor [Cp*CoCl2]2 with 2 equivalents of the corresponding N,O-type ligand in the presence of base at ambient temperatures (Scheme [13]).[36] The resulting cobalt complexes were found to be air-stable, thus their preparations could conveniently be operated under atmospheric conditions and avoid the need for glovebox. Given that the catalytic activity is closely related to the facile formation and delivery of electrophilic metal-nitrenoid species,[24] electronic properties of κ2-N,O chelating ligands were finely tuned by changing substituents in N,O-ligands. We found that 8-hydroxylquinoline ligands with electron-withdrawing groups were highly facile in the desired C–H amidation. Among those, Co1 bearing 5,7-dichloro quinolinol ligand turned out to be the most efficient in the catalytic outer-sphere C–N bond-forming reactions investigated below (vide infra).

C(sp2)–N Bond Formation
Cyclic carbamates are a key structural motif not only present in numerous medicinally active compounds,[37] but also utilized as a useful functional handle in chemical synthesis (e.g., as a chiral auxiliary).[38] Despite their importance, metal-nitrenoid-based C(sp2)–H amidation of arenes was rarely applied to the synthesis of benzo-fused cyclic carbamates, mainly due to the difficulty in controlling the regioselectivity.[39] In this context, we explored the utility of Cp*Co(LX) catalyst (Co1) in the C(sp2)–H amidation of phenyl azidoformates, harnessing the azide motif as a nitrene precursor (Scheme [14]).[36] The customized catalyst Co1 indeed led to the desired intramolecular C–H amidation of various types of aryl azidoformates to give five-membered cyclic carbamates, indicating that the Cp*Co(N,O)-type system is suitable for outer-sphere C–H amidation pathway. When benzyl azidoformate was subjected, the spirocyclization and subsequent skeletal rearrangement (C–C migration) took place to afford six-membered cyclic carbamate.

With the optimized Co1 system, high regioselectivity (C6/C2 amidation) was achieved in the C–H amidation of meta-substituted phenyl azidoformates (Scheme [15a]). It was of our special interest to observe that the regioselectivity was modulated according to the central metal within the same ligand system. For instance, an iridium analogue (Ir1) containing an identical ligand as that of Co1 was also able to promote the desired C–H amidation with similar efficiency but with reduced C6/C2 selectivity. To understand the contrasting regioselectivity, we briefly compared the structural features of the two metal complexes by X-ray crystallographic analysis of the solid structures of Co1 and Ir1 (Scheme [15b]). The distances between the Co metal center and the coordinating ligands (center of the Cp* plane, N, and O atoms) in Co1 were noticeably shorter than those of Ir1. This is presumably due to the smaller ionic metal radius of cobalt than iridium. Considering the previous report that the smaller ionic metal radius of cobalt rendered Cp*Co more susceptible to steric perturbations,[7] we postulated that the distinct structural features of Co1 lead to favoring the insertion of key metal-nitrenoid intermediates at the less-hindered C6 position.
The invention of the Cp*Co(N,O) system for outer-sphere C(sp2)–H amidation inspired us to explore further utility of cobalt-nitrenoid transfer into more specialized aromatic systems. Given that arenium species bearing a quaternary carbon center were known to stimulate alkyl migration,[40] we speculated that the insertion of Co-nitrenoid into alkyl-substituted aromatic carbon would generate the arenium species and trigger C–C rearrangement. When 2,6-dialkyl-substiuted phenyl azidoformate was employed, the Cp*Co catalyst (Co1) exhibited unconventional amidation reactivity accompanying an intriguing [1,2]-relocation of the isopropyl or benzyl group (Scheme [16]).[41] This cascade strategy represents the first example of alkyl migration reactions mediated by Co-nitrenoid insertion into aromatic systems. When the cobalt system was applied to the linear ethyl-substituted substrate, [1,2]-ethyl rearrangement, as well as [1,4]-ethyl shift took place to give the regioisomeric amidation products.


Mulliken spin density analysis suggested that the key cationic arenium species A is indeed generated after the cobalt-nitrenoid insertion to the alkyl-substituted ortho-carbon, considering that the spin is localized at the cobalt metal center (Scheme [17a]). For the first time, an ‘alkyl-walking mechanism was proposed as a mechanistic mode for the [1,4]-carbon relocation, wherein the key complex A experiences the migration in sequence by traversing TS1, TS2, and TS3 with reasonable barriers (Scheme [17b]). The mechanistic scenario suggested by the computational simulation was strongly supported by an experimental observation of formal cyclohexyl migration (Scheme [17c]). The formal cyclohexyl relocation accompanying with [1,3]-ethyl shifts can best be rationalized by the intervention of the alkyl-walking and subsequent spirocyclization rearrangements,[42] thereby this finding provides a strong evidence for the novel mechanism.

While nonafluoro-tert-butanol (NFTB) solvent was effective for the above-mentioned amidative alkyl migration, the use of hexafluoro-isopropanol (HFIP) as solvent completely changed the chemical reactivity (Scheme [18]).[43] In HFIP solvent system, subjecting 2,6-dimethyl phenyl azidoformate to the identical Co1 catalytic conditions afforded HFIP-incorporated bisamidated endo-cyclodimers resulting from Diels–Alder dimerization. Interestingly, additional alkyl alcohols could also be introduced into the dimeric Diels–Alder scaffold when they were used in a co-solvent system containing NFTB. In this manner, a range of alcohols such as methyl, allyl, or benzyl moieties could be embedded to furnish the corresponding Meoc-, Alloc-, and Cbz-equipped dimeric products.


Next, DFT calculations were conducted to elucidate the underlying mechanism of Diels–Alder dimerization with the choice of the specific alcoholic solvents (Scheme [19]). Quantum-chemical calculations indicated that the ring opening of arenium carbamate E is efficient to generate the key ortho-quinamine scaffold F. The o-quinamine F is then considered to undergo alcohol incorporation for subsequent Diels–Alder dimerization. The insertion of HFIP to the structure F (12.4 kcal/mol) was calculated to be energetically more feasible than the competing alkyl migration path (15.5 kcal/mol), thus proceeding with following Diels–Alder dimerization to give dimeric products. On the other hand, the corresponding NFTB incorporation (19.3 kcal/mol) is energetically much more demanding, thus the alkyl migration becomes more favored in NFTB solvent system.
Additionally, this intriguing amidative Diels–Alder dimerization reaction was coupled with light irradiation to provoke further structural diversity in the bisamidated endo-cyclodimer (Scheme [20]). In the presence of UV light (365 nm), a ring-fused cage compound was obtained in high yield from [2+2] cycloaddition of the dimeric product. It should be highlighted that this simple cobalt-based catalytic system enabled the transformation of aromatic scaffold into a dearomatized and fully saturated cage structure loaded with bisamides.

# 3.2
C(sp3)–N Bond Formation
As a natural extension, we wondered whether the present Co-nitrenoid catalytic system could also be applied to C(sp3)–H amidation reactions. This premise was implemented in a model reaction with phenethyl azidoformate under the same catalytic system with Co1 (Scheme [21]).[36] The cyclization was highly selective for the sp 3-benzylic site, and a competing C(sp2)–H amidation was not observed. The cobalt catalyst Co1 was also effective in amidation of aliphatic substrates containing tertiary, secondary, and primary C–H bonds, providing the corresponding oxazolidinones. In terms of nitrenoid insertion to the C–H bond α to the heteroatom, the Co1 system was much more efficient than the Ir1 system, demonstrating another interesting feature that can be achieved by altering the central metal within the same ligand environment.

Next, we turned our attention to extending the application of the Cp*Co-nitrenoid catalytic platform from an intramolecular manner to an intermolecular version, particularly to unactivated alkanes containing inert C–H bonds. To tackle the challenging alkane C–H bonds, the electronic property of the (N,O)-type ligands of cobalt catalysts was modulated by introducing a range of substituents (Scheme [22]).[44] In a model reaction of n-hexane and 2,2,2-trichloroethoxycarbonyl azide (TrocN3) as a nitrene precursor, cobalt catalysts with ligands containing electron-withdrawing groups, especially a 5-nitro group (Co2), turned out to be optimal for catalytic intermolecular C–H amidation.

Frontier molecular orbital analysis of this reaction suggested that β-LUMO+2 of Co-nitrenoid plays a crucial role in abstracting a hydrogen atom from alkanes and thus is closely related to the catalytic amidation efficiency. In this respect, the computations provided a corroborative trend that the stronger the electron-withdrawing property present in the (N,O)-ligand of cobalt catalyst, the lower the β-LUMO+2 energy of the cobalt nitrenoid, indicating that electronic perturbation at the Co-nitrenoid indeed alter the amidation reactivity.
The proposed mechanism of the outer-sphere C–H amidation of the unactivated alkanes is outlined in Scheme [23]. Upon TrocN3 coordination to cobalt, the resultant adduct XI undergoes nitrogen extrusion to engender the key triplet cobalt-nitrenoid XII. Subsequently, the radical-type hydrogen atom abstraction/radical rebound pathway was suggested to operate on the basis the EPR and radical clock experimental results along with the consistent computational results.

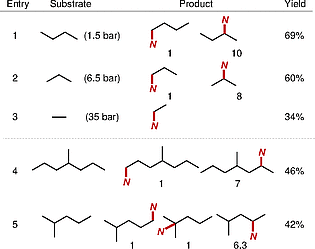
With the newly optimized cobalt catalyst Co2, we evaluated the reaction efficiency and site selectivity in the intermolecular C(sp3)–H amidation of a range of readily available unactivated hydrocarbon feedstock (Table [1]). The cobalt catalytic system was found to be highly facile for the intermolecular amidation of light alkanes, such as n-butane, propane, and ethane in a standard pressure reactor. More complex alkanes bearing multiple carbon chains were also successfully amidated under the Cp*Co catalytic system to provide isomeric mixtures of amidation products in moderate yield.
In terms of site selectivity, Co2 catalytic system offered a high degree of secondary C–H bond selectivity in the presence of primary or tertiary C–H bonds. Given that the C–H functionalization involved in the radical-type hydrogen atom abstraction (HAA) mechanism tends to favor tertiary C–H bonds over secondary C–H bonds due to lower bond-dissociation energies, the secondary C–H bond preference can be attributed to the catalyst-mediated site selectivity, reversing the intrinsic tertiary C–H selectivity. We rationalized that the collective two-point steric interaction of two ligands (Cp* and N,O-ligand)[33] appears to impose difficulties accessing the sterically hindered tertiary C–H bonds.
#
# 4
Conclusion and Outlook
In this Account, we have summarized our research efforts toward utilization of Cp*Co(III) catalysts in a broad range of C–N bond-forming reactions, featuring high reaction efficiency and site selectivity. This cobalt catalytic system can adopt two distinct mechanistic reaction pathways to forge C–N bonds: the inner-sphere and the outer-sphere pathway. The inner-sphere C–H amidation of arenes was enabled via chelation-assisted regiocontrol with the utilization of O-acylcarbamates and dioxazolones as convenient amidating sources. To enable outer-sphere C–H amidation, we developed for the first time a Cp*Co(III)(κ2-N,O chelate) complex. This catalytic system allowed an efficient transfer of putative cobalt-nitrenoids into a wide range of C(sp 2)–H and C(sp 3)–H bonds to produce various types of five- or six-membered cyclic carbamates.
Despite the advances of Cp*Co(III)-catalyzed C–H amidation reactions, several issues still remain for future research directions. One of the major challenges is the successful implementation of the Cp*Co catalytic system in the late-stage functionalization of complex molecules, which requires the further endeavors to develop site- and/or chemoselective C–H functionalization protocols. Next, although Cp*Co(III)-catalyzed outer-sphere amidation have been investigated with both activated and unactivated C(sp3)–H bonds, the reactivity of these systems have been mainly limited to racemic systems. Thus, the development of enantioselective outer-sphere C(sp3)–H amination reaction is highly desirable. The integration of Cp*Co(III) catalysts with photocatalysis or electrocatalysis to establish novel oxidative C–H functionalization would be also a promising research subject, especially from a sustainable perspective. In addition, efforts should be made to employ alternative nitrene precursors in addition to azidoformates in the outer-sphere C–N bond-forming reactions to produce more diverse types of amino functional groups. The application of carbene precursors will also be an interesting research focus for the efficient C–C bond formation utilizing the current Cp*Co(III) catalytic system.
We believe that Cp*Co(III) complexes, characterized by inexpensive and abundant 3d-metal-based catalyst with the readily tunable ligand system, will serve as an attractive catalytic platform to achieve further exciting breakthroughs in the relevant C–H functionalization reactions. We hope this Account will provide a concise overview of Cp*Co(III) catalytic system for synthetic elaboration entailed to reach other methodological advances.
#
#
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
This Account is dedicated to Professor Masahiro Murakami on his retirement in honor of his incalculable contributions to chemistry and to the chemical community. The authors thank to Dr. Sangwon Seo for helpful discussions and critical reading.
-
References
- 1a Davies HM, Morton D. J. Org. Chem. 2016; 81: 343
- 1b Rogge T, Kaplaneris N, Chatani N, Kim J, Chang S, Punji B, Schafer LL, Musaev DG, Wencel-Delord J, Roberts CA, Sarpong R, Wilson ZE, Brimble MA, Johansson MJ, Ackermann L. Nat. Rev. Methods Primers 2021; 1: 43
- 2a Meng G, Lam NY. S, Lucas EL, Saint-Denis TG, Verma P, Chekshin N, Yu J.-Q. J. Am. Chem. Soc. 2020; 142: 10571
- 2b Zhang C, Li Z.-L, Gu Q.-S, Liu X.-Y. Nat. Commun. 2021; 12: 475
- 3a Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007; 36: 1173
- 3b Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L. Chem. Rev. 2019; 119: 2192
- 3c Kim K, Cho S, Park S, Lee Y. Bull. Korean Chem. Soc. 2021; 42: 699
- 3d Kim TK, Youn SW. Bull. Korean Chem. Soc. 2021; 42: 521
- 3e Lee YL, Lee KR, Xuan Z, Lee S.-g. Bull. Korean Chem. Soc. 2021; 42: 537
- 3f Barranco S, Zhang J, López-Resano S, Casnati A, Pérez-Temprano MH. Nat. Synth. 2022; 1: 841
- 4a Piou T, Rovis T. Acc. Chem. Res. 2018; 51: 170
- 4b Li X, Ouyang W, Nie J, Ji S, Chen Q, Huo Y. ChemCatChem 2020; 12: 2358
- 4c Kim J, Jin S, Kim D, Chang S. Bull. Korean Chem. Soc. 2021; 42: 529
- 5 Yoshino T, Ikemoto H, Matsunaga S, Kanai M. Angew. Chem. Int. Ed. 2013; 52: 2207
- 6 Park J, Chang S. Chem. Asian J. 2018; 13: 1089
- 7 Wang X, Lerchen A, Glorius F. Org. Lett. 2016; 18: 2090
- 8 Lu H, Hu Y, Jiang H, Wojtas L, Zhang XP. Org. Lett. 2012; 14: 5158
- 9 Yu D.-G, Gensch T, de Azambuja F, Vásquez-Céspedes S, Glorius F. J. Am. Chem. Soc. 2014; 136: 17722
- 10a Shaaban S, Davies C, Waldmann H. Eur. J. Org. Chem. 2020; 6512
- 10b Yoshino T, Matsunaga S. ACS Catal. 2021; 11: 6455
- 11 Pesciaioli F, Dhawa U, Oliveira JC, Yin R, John M, Ackermann L. Angew. Chem. Int. Ed. 2018; 57: 15425
- 12 Ozols K, Jang Y.-S, Cramer N. J. Am. Chem. Soc. 2019; 141: 5675
- 13a Huang Y, Pi C, Tang Z, Wu Y, Cui X. Chin. Chem. Lett. 2020; 31: 3237
- 13b Yan R, Yu H, Wang Z.-X. Chin. J. Chem. 2021; 39: 1205
- 13c Yu Y, Xia Z, Wu Q, Liu D, Yu L, Xiao Y, Tan Z, Deng W, Zhu G. Chin. Chem. Lett. 2021; 32: 1263
- 14 Wang S, Chen S.-Y, Yu X.-Q. Chem. Commun. 2017; 53: 3165
- 15 Mei R, Dhawa U, Samanta RC, Ma W, Wencel-Delord J, Ackermann L. ChemSusChem 2020; 13: 3306
- 16a Yoshino T, Matsunaga S. Adv. Synth. Catal. 2017; 359: 1245
- 16b Yoshino T, Matsunaga S. Synlett 2019; 30: 1384
- 16c Yoshino T, Satake S, Matsunaga S. Chem. Eur. J. 2020; 26: 7346
- 17 Sun B, Yoshino T, Matsunaga S, Kanai M. Adv. Synth. Catal. 2014; 356: 1491
- 18 Patel P, Chang S. ACS Catal. 2015; 5: 853
- 19a Park Y, Jee S, Kim JG, Chang S. Org. Process Res. Dev. 2015; 19: 1024
- 19b Park Y, Park KT, Kim JG, Chang S. J. Am. Chem. Soc. 2015; 137: 4534
- 20 Park J, Chang S. Angew. Chem. Int. Ed. 2015; 54: 14103
- 21 Tan PW, Mak AM, Sullivan MB, Dixon DJ, Seayad J. Angew. Chem. Int. Ed. 2017; 56: 16550
- 22 Fukagawa S, Kato Y, Tanaka R, Kojima M, Yoshino T, Matsunaga S. Angew. Chem. Int. Ed. 2019; 58: 1153
- 23 Breslow R, Gellman SH. J. Chem. Soc., Chem. Commun. 1982; 1400
- 24 Park Y, Kim Y, Chang S. Chem. Rev. 2017; 117: 9247
- 25a Kim JY, Park SH, Ryu J, Cho SH, Kim SH, Chang S. J. Am. Chem. Soc. 2012; 134: 9110
- 25b Ryu J, Shin K, Park SH, Kim JY, Chang S. Angew. Chem. Int. Ed. 2012; 51: 9904
- 25c Ryu J, Kwak J, Shin K, Lee D, Chang S. J. Am. Chem. Soc. 2013; 135: 12861
- 25d Shin K, Baek Y, Chang S. Angew. Chem. Int. Ed. 2013; 52: 8031
- 25e Hwang H, Kim J, Jeong J, Chang S. J. Am. Chem. Soc. 2014; 136: 10770
- 25f Kim J, Chang S. Angew. Chem. Int. Ed. 2014; 53: 2203
- 25g Park SH, Kwak J, Shin K, Ryu J, Park Y, Chang S. J. Am. Chem. Soc. 2014; 136: 2492
- 25h Shin K, Ryu J, Chang S. Org. Lett. 2014; 16: 2022
- 25i Shin K, Kim H, Chang S. Acc. Chem. Res. 2015; 48: 1040
- 25j Park Y, Heo J, Baik M.-H, Chang S. J. Am. Chem. Soc. 2016; 138: 14020
- 25k Hwang Y, Park Y, Chang S. Chem. Eur. J. 2017; 23: 11147
- 25l Park J, Lee J, Chang S. Angew. Chem. Int. Ed. 2017; 56: 4256
- 26 Hong SY, Park Y, Hwang Y, Kim YB, Baik M.-H, Chang S. Science 2018; 359: 1016
- 27 Hong SY, Hwang Y, Lee M, Chang S. Acc. Chem. Res. 2021; 54: 2683
- 28a Park Y, Chang S. Nat. Catal. 2019; 2: 219
- 28b Hong SY, Kim D, Chang S. Nat. Catal. 2021; 4: 79
- 28c Kim S, Kim D, Hong SY, Chang S. J. Am. Chem. Soc. 2021; 143: 3993
- 28d Lee E, Hwang Y, Kim YB, Kim D, Chang S. J. Am. Chem. Soc. 2021; 143: 6363
- 29 Hwang Y, Park Y, Kim YB, Kim D, Chang S. Angew. Chem. Int. Ed. 2018; 57: 13565
- 30 Kweon J, Kim D, Kang S, Chang S. J. Am. Chem. Soc. 2022; 144: 1872
- 31 Hong SY, Son J, Kim D, Chang S. J. Am. Chem. Soc. 2018; 140: 12359
- 32 Hwang Y, Baek SB, Kim D, Chang S. J. Am. Chem. Soc. 2022; 144: 4277
- 33 Hwang Y, Jung H, Lee E, Kim D, Chang S. J. Am. Chem. Soc. 2020; 142: 8880
- 34 Jung H, Schrader M, Kim D, Baik M.-H, Park Y, Chang S. J. Am. Chem. Soc. 2019; 141: 15356
- 35a Lee M, Jung H, Kim D, Park J.-W, Chang S. J. Am. Chem. Soc. 2020; 142: 11999
- 35b Gwon Y, Lee M, Kim D, Chang S. Org. Lett. 2022; 24: 1088
- 36 Lee J, Lee J, Jung H, Kim D, Park J, Chang S. J. Am. Chem. Soc. 2020; 142: 12324
- 37 Niemi T, Repo T. Eur. J. Org. Chem. 2019; 1180
- 38 Heravi MM, Zadsirjan V. Tetrahedron: Asymmetry 2013; 24: 1149
- 39 Jiao J, Murakami K, Itami K. ACS Catal. 2016; 6: 610
- 40a Allen RH, Alfrey TJr, Yats LD. J. Am. Chem. Soc. 1959; 81: 42
- 40b Olah GA, Olah JA, Ohyama T. J. Am. Chem. Soc. 1984; 106: 5284
- 41 Lee J, Kang B, Kim D, Lee J, Chang S. J. Am. Chem. Soc. 2021; 143: 18406
- 42 Woodward R, Singh T. J. Am. Chem. Soc. 1950; 72: 494
- 43 Lee J, Kang B, Kim D, Chang S. Org. Lett. 2022; 24: 5845
- 44 Lee J, Jin S, Kim D, Hong SH, Chang S. J. Am. Chem. Soc. 2021; 143: 5191
Corresponding Author
Publikationsverlauf
Eingereicht: 31. Oktober 2022
Angenommen nach Revision: 24. November 2022
Accepted Manuscript online:
25. November 2022
Artikel online veröffentlicht:
11. Januar 2023
© 2022. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1a Davies HM, Morton D. J. Org. Chem. 2016; 81: 343
- 1b Rogge T, Kaplaneris N, Chatani N, Kim J, Chang S, Punji B, Schafer LL, Musaev DG, Wencel-Delord J, Roberts CA, Sarpong R, Wilson ZE, Brimble MA, Johansson MJ, Ackermann L. Nat. Rev. Methods Primers 2021; 1: 43
- 2a Meng G, Lam NY. S, Lucas EL, Saint-Denis TG, Verma P, Chekshin N, Yu J.-Q. J. Am. Chem. Soc. 2020; 142: 10571
- 2b Zhang C, Li Z.-L, Gu Q.-S, Liu X.-Y. Nat. Commun. 2021; 12: 475
- 3a Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007; 36: 1173
- 3b Gandeepan P, Müller T, Zell D, Cera G, Warratz S, Ackermann L. Chem. Rev. 2019; 119: 2192
- 3c Kim K, Cho S, Park S, Lee Y. Bull. Korean Chem. Soc. 2021; 42: 699
- 3d Kim TK, Youn SW. Bull. Korean Chem. Soc. 2021; 42: 521
- 3e Lee YL, Lee KR, Xuan Z, Lee S.-g. Bull. Korean Chem. Soc. 2021; 42: 537
- 3f Barranco S, Zhang J, López-Resano S, Casnati A, Pérez-Temprano MH. Nat. Synth. 2022; 1: 841
- 4a Piou T, Rovis T. Acc. Chem. Res. 2018; 51: 170
- 4b Li X, Ouyang W, Nie J, Ji S, Chen Q, Huo Y. ChemCatChem 2020; 12: 2358
- 4c Kim J, Jin S, Kim D, Chang S. Bull. Korean Chem. Soc. 2021; 42: 529
- 5 Yoshino T, Ikemoto H, Matsunaga S, Kanai M. Angew. Chem. Int. Ed. 2013; 52: 2207
- 6 Park J, Chang S. Chem. Asian J. 2018; 13: 1089
- 7 Wang X, Lerchen A, Glorius F. Org. Lett. 2016; 18: 2090
- 8 Lu H, Hu Y, Jiang H, Wojtas L, Zhang XP. Org. Lett. 2012; 14: 5158
- 9 Yu D.-G, Gensch T, de Azambuja F, Vásquez-Céspedes S, Glorius F. J. Am. Chem. Soc. 2014; 136: 17722
- 10a Shaaban S, Davies C, Waldmann H. Eur. J. Org. Chem. 2020; 6512
- 10b Yoshino T, Matsunaga S. ACS Catal. 2021; 11: 6455
- 11 Pesciaioli F, Dhawa U, Oliveira JC, Yin R, John M, Ackermann L. Angew. Chem. Int. Ed. 2018; 57: 15425
- 12 Ozols K, Jang Y.-S, Cramer N. J. Am. Chem. Soc. 2019; 141: 5675
- 13a Huang Y, Pi C, Tang Z, Wu Y, Cui X. Chin. Chem. Lett. 2020; 31: 3237
- 13b Yan R, Yu H, Wang Z.-X. Chin. J. Chem. 2021; 39: 1205
- 13c Yu Y, Xia Z, Wu Q, Liu D, Yu L, Xiao Y, Tan Z, Deng W, Zhu G. Chin. Chem. Lett. 2021; 32: 1263
- 14 Wang S, Chen S.-Y, Yu X.-Q. Chem. Commun. 2017; 53: 3165
- 15 Mei R, Dhawa U, Samanta RC, Ma W, Wencel-Delord J, Ackermann L. ChemSusChem 2020; 13: 3306
- 16a Yoshino T, Matsunaga S. Adv. Synth. Catal. 2017; 359: 1245
- 16b Yoshino T, Matsunaga S. Synlett 2019; 30: 1384
- 16c Yoshino T, Satake S, Matsunaga S. Chem. Eur. J. 2020; 26: 7346
- 17 Sun B, Yoshino T, Matsunaga S, Kanai M. Adv. Synth. Catal. 2014; 356: 1491
- 18 Patel P, Chang S. ACS Catal. 2015; 5: 853
- 19a Park Y, Jee S, Kim JG, Chang S. Org. Process Res. Dev. 2015; 19: 1024
- 19b Park Y, Park KT, Kim JG, Chang S. J. Am. Chem. Soc. 2015; 137: 4534
- 20 Park J, Chang S. Angew. Chem. Int. Ed. 2015; 54: 14103
- 21 Tan PW, Mak AM, Sullivan MB, Dixon DJ, Seayad J. Angew. Chem. Int. Ed. 2017; 56: 16550
- 22 Fukagawa S, Kato Y, Tanaka R, Kojima M, Yoshino T, Matsunaga S. Angew. Chem. Int. Ed. 2019; 58: 1153
- 23 Breslow R, Gellman SH. J. Chem. Soc., Chem. Commun. 1982; 1400
- 24 Park Y, Kim Y, Chang S. Chem. Rev. 2017; 117: 9247
- 25a Kim JY, Park SH, Ryu J, Cho SH, Kim SH, Chang S. J. Am. Chem. Soc. 2012; 134: 9110
- 25b Ryu J, Shin K, Park SH, Kim JY, Chang S. Angew. Chem. Int. Ed. 2012; 51: 9904
- 25c Ryu J, Kwak J, Shin K, Lee D, Chang S. J. Am. Chem. Soc. 2013; 135: 12861
- 25d Shin K, Baek Y, Chang S. Angew. Chem. Int. Ed. 2013; 52: 8031
- 25e Hwang H, Kim J, Jeong J, Chang S. J. Am. Chem. Soc. 2014; 136: 10770
- 25f Kim J, Chang S. Angew. Chem. Int. Ed. 2014; 53: 2203
- 25g Park SH, Kwak J, Shin K, Ryu J, Park Y, Chang S. J. Am. Chem. Soc. 2014; 136: 2492
- 25h Shin K, Ryu J, Chang S. Org. Lett. 2014; 16: 2022
- 25i Shin K, Kim H, Chang S. Acc. Chem. Res. 2015; 48: 1040
- 25j Park Y, Heo J, Baik M.-H, Chang S. J. Am. Chem. Soc. 2016; 138: 14020
- 25k Hwang Y, Park Y, Chang S. Chem. Eur. J. 2017; 23: 11147
- 25l Park J, Lee J, Chang S. Angew. Chem. Int. Ed. 2017; 56: 4256
- 26 Hong SY, Park Y, Hwang Y, Kim YB, Baik M.-H, Chang S. Science 2018; 359: 1016
- 27 Hong SY, Hwang Y, Lee M, Chang S. Acc. Chem. Res. 2021; 54: 2683
- 28a Park Y, Chang S. Nat. Catal. 2019; 2: 219
- 28b Hong SY, Kim D, Chang S. Nat. Catal. 2021; 4: 79
- 28c Kim S, Kim D, Hong SY, Chang S. J. Am. Chem. Soc. 2021; 143: 3993
- 28d Lee E, Hwang Y, Kim YB, Kim D, Chang S. J. Am. Chem. Soc. 2021; 143: 6363
- 29 Hwang Y, Park Y, Kim YB, Kim D, Chang S. Angew. Chem. Int. Ed. 2018; 57: 13565
- 30 Kweon J, Kim D, Kang S, Chang S. J. Am. Chem. Soc. 2022; 144: 1872
- 31 Hong SY, Son J, Kim D, Chang S. J. Am. Chem. Soc. 2018; 140: 12359
- 32 Hwang Y, Baek SB, Kim D, Chang S. J. Am. Chem. Soc. 2022; 144: 4277
- 33 Hwang Y, Jung H, Lee E, Kim D, Chang S. J. Am. Chem. Soc. 2020; 142: 8880
- 34 Jung H, Schrader M, Kim D, Baik M.-H, Park Y, Chang S. J. Am. Chem. Soc. 2019; 141: 15356
- 35a Lee M, Jung H, Kim D, Park J.-W, Chang S. J. Am. Chem. Soc. 2020; 142: 11999
- 35b Gwon Y, Lee M, Kim D, Chang S. Org. Lett. 2022; 24: 1088
- 36 Lee J, Lee J, Jung H, Kim D, Park J, Chang S. J. Am. Chem. Soc. 2020; 142: 12324
- 37 Niemi T, Repo T. Eur. J. Org. Chem. 2019; 1180
- 38 Heravi MM, Zadsirjan V. Tetrahedron: Asymmetry 2013; 24: 1149
- 39 Jiao J, Murakami K, Itami K. ACS Catal. 2016; 6: 610
- 40a Allen RH, Alfrey TJr, Yats LD. J. Am. Chem. Soc. 1959; 81: 42
- 40b Olah GA, Olah JA, Ohyama T. J. Am. Chem. Soc. 1984; 106: 5284
- 41 Lee J, Kang B, Kim D, Lee J, Chang S. J. Am. Chem. Soc. 2021; 143: 18406
- 42 Woodward R, Singh T. J. Am. Chem. Soc. 1950; 72: 494
- 43 Lee J, Kang B, Kim D, Chang S. Org. Lett. 2022; 24: 5845
- 44 Lee J, Jin S, Kim D, Hong SH, Chang S. J. Am. Chem. Soc. 2021; 143: 5191