
Subscribe to RSS
Please copy the URL and add it into your RSS Feed Reader.
https://www.thieme-connect.de/rss/thieme/en/10.1055-s-00000083.xml
Synlett 2016; 27(15): 2201-2204
DOI: 10.1055/s-0035-1562725
DOI: 10.1055/s-0035-1562725
letter
Lithium Sulfondiimidoyl Carbanions
Further Information
Publication History
Received: 23 May 2016
Accepted: 10 June 2016
Publication Date:
13 July 2016 (online)
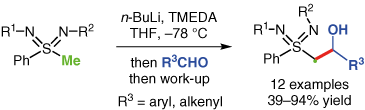

Abstract
The trapping of sulfondiimine-based α-lithiated carbanions with aldehydes leads to unprecedented β-hydroxy sulfondiimines in good yields. An alkylation has been achieved by treatment of the metalated species with methyl iodide. Of special interest, N-monosubstituted β-hydroxy sulfondiimines can be accessed without protection of the free nitrogen.
Supporting Information
- Supporting information for this article is available online at http://dx.doi.org/10.1055/s-0035-1562725.
- Supporting Information
-
References and Notes
- 1a Lücking U. Angew. Chem. Int. Ed. 2013; 52: 9399 ; and references cited therein
- 1b Lücking U, Scholz A, Lienau P, Siemeister G, Bohlmann R, Bömer U. WO 2015150273, 2015
- 1c Sparks TC, Loso MR, Watson GB, Babcock JM, Kramer VJ, Zhu Y, Nugent BM, Thomas JD In Modern Crop Protection Compounds . Kraemer W, Schirmer U, Jeschke P, Witschel M. Wiley-VCH; Weinheim: 2012: 1226
- 2a Johnson CR. Acc. Chem. Res. 1973; 6: 341
- 2b Pyne S. Sulfur Rep. 1992; 12: 57
- 2c Reggelin M, Zur C. Synthesis 2000; 1
- 2d Gais H.-J. Heteroat. Chem. 2007; 18: 472
- 3a Harmata M. Chemtracts 2003; 16: 660
- 3b Okamura H, Bolm C. Chem. Lett. 2004; 32: 482
- 3c Bolm C In Asymmetric Synthesis with Chemical and Biological Methods . Enders D, Jäger K.-E. Wiley-VCH; Weinheim: 2007: 149
- 4a Johnson CR, Shanklin JR, Kirchhoff RA. J. Am. Chem. Soc. 1973; 95: 6462
- 4b Johnson CR, Stark CJ. Tetrahedron Lett. 1979; 49: 4713
- 4c Johnson CR, Kirchhoff RA. J. Am. Chem. Soc. 1979; 101: 3602
- 4d Johnson CR, Zeller JR. J. Am. Chem. Soc. 1982; 104: 4021
- 4e Johnson CR, Barbachyn MR. J. Am. Chem. Soc. 1982; 104: 4290
- 4f Bolm C, Seger A, Felder M. Tetrahedron Lett. 1993; 34: 8079
- 4g Sedelmeier J, Bolm C. J. Org. Chem. 2007; 72: 8859
- 5a Candy M, Guyon C, Mersmann S, Chen J.-R, Bolm C. Angew. Chem. Int. Ed. 2012; 51: 4440
- 5b Candy M, Bohmann RA, Bolm C. Adv. Synth. Catal. 2012; 354: 2928
- 5c Bohmann RA, Bolm C. Org. Lett. 2013; 15: 4277
- 5d Hendriks CM. M, Bohmann RA, Bohlem M, Bolm C. Adv. Synth. Catal. 2014; 356: 1847
- 5e Bohmann RA, Unoh Y, Miura M, Bolm C. Chem. Eur. J. 2016; 22: 6783
- 6a Diederich WE, Haake M. J. Org. Chem. 2003; 68: 3817
- 6b Dehli J, Bolm C. Synthesis 2005; 1058
- 6c Yoshimura T, Fujie T, Fujii T. Tetrahedron Lett. 2007; 48: 427
- 7 The attempt to deprotonate 2a with 2.0 equiv of the superbase generated from KOH in DMSO (at 20 °C for 15 min followed by treatment with 5a at the same temperature), which recently proved effective in N-alkylations of NH-sulfondiimines (comp. ref. 5d), remained unsuccessful coming along with partial decomposition of 2a.
- 8 General Experimental Procedure Under an atmosphere of argon, in a flame-dried Schlenk tube equipped with magnetic stirrer and septum, the sulfondiimine (1.0 equiv, 0.163 mmol) was dissolved in dry THF (2 mL). TMEDA (2.0 equiv) was added, and the resulting solution was cooled to –78 °C. Then, n-BuLi (1.05 equiv or 2.10 equiv, depending on the substrate) was added dropwise at –78 °C within 5 min, and the resulting solution was stirred for 30 min at –78 °C. The electrophilic reagent (see each case for details) was then added by syringe, and the reaction mixture was stirred at –78 °C. The conversion of the reaction was monitored by TLC. In case of an incomplete conversion, the reaction mixture was slowly warmed to room temperature (see each case for details). Upon complete conversion sat. NH4Cl solution (15 mL) was added. The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous MgSO4, and the solvents were removed under reduced pressure. Purification by flash column chromatography using n-pentane–EtOAc gradient elution (see each case for details) afforded the desired product.
- 9 Analytical Data for Selected Products 1-(N,N′-Diphenyl-S-phenylsulfondiimidoyl)-2-phenylethan-2-ol (3a) 94% yield. 1H NMR (600 MHz, CDCl3): δ = 8.24 (d, J = 7.7 Hz, 2 H), 7.69–7.65 (m, 1 H), 7.63–7.58 (m, 2 H), 7.27–7.14 (m, 13 H), 7.02 (br s, 1 H, OH), 7.96–7.91 (m, 2 H), 5.00 (d, J = 10.4 Hz, 1 H), 4.00 (dd, J = 14.1, 10.4 Hz, 1 H), 2.99 (d, J = 14.1 Hz, 1 H). 13C NMR (151 MHz, CDCl3): δ = 144.6, 114.4, 140.9, 136.4, 133.5, 129.8 (2 C), 129.3 (2 C), 129.2 (2 C), 129.2 (2 C), 128.6 (2 C), 128.0, 125.6 (2 C), 123.4 (2 C), 122.8 (2 C), 122.1, 121.8, 69.2, 62.8. MS (EI): m/z (%) = 412 (6) [M]+, 320 (15), 199 (100), 197 (33), 181 (22), 180 (25), 167 (31), 165 (45), 123 (29), 106 (22), 105 (24), 103 (27), 93 (17), 77 (41). IR (neat): ν = 3269, 3061, 2863, 1590, 1483, 1446, 1284, 1247, 1176, 1068, 974, 906, 856, 832, 792, 747, 690 cm–1. ESI-HRMS: m/z calcd for C26H25ON2S: 413.1682; found: 413.1682. N,N′-Diphenyl-S-ethyl-S-phenyl-sulfondiimine (7) 94% yield; mp 125–126 °C. 1H NMR (600 MHz, CDCl3): δ = 8.21–8.28 (m, 2 H), 7.61–7.52 (m, 3 H), 7.20–7.12 (m, 8 H), 6.88–6.84 (m, 2 H), 3.64 (q, J = 7.4 Hz, 2 H), 1.07 (t, J = 7.4 Hz, 3 H). 13C NMR (151 MHz, CDCl3): δ = 145.9 (2 C), 136.1, 132.8, 129.5 (2 C), 129.4 (2 C), 129.1 (4 C), 123.0 (4 C), 121.0 (2 C), 50.2, 7.8. MS (EI): m/z (%) = 320 (26) [M]+, 229 (59), 200 (100), 182 (20), 167 (17), 105 (10), 97 (17), 77 (15). IR (KBr): ν = 3054, 1590, 1480, 1445, 1285, 1251, 1174, 1067, 1023, 964, 797, 747, 692. ESI-HRMS: m/z calcd for C20H21N2S: 321.1420; found: 321.1420.
- 10 To our surprise, neither acetone nor acetophenone (2.0 equiv in each case) could be employed as electrophile. Even after 48 h at 20 °C no addition product of type 3 was observed. Only partial degradation of 2a had occurred.
- 11 Here, both 2b and 1a were applied as racemates. For accessing enantiopure sulfondiimines, see ref. 5a and references cited therein.
- 12 Attempts to separate the diastereomers of 3k remained unsuccessful.
- 13a Hwang K.-J, Logusch EW, Brannigan LH, Thomson MR. J. Org. Chem. 1987; 52: 3435
- 13b Bolm C, Felder M. Tetrahedron Lett. 1993; 34: 6041
- 13c Bolm C, Felder M. Synlett 1994; 655
- 13d Kawanishi H, Morimoto H, Nakano T, Miyajima T, Oda K, Takeda K, Yano S, Hirano N, Tsujihara K. Heterocycles 1998; 49: 169
- 13e Shen X, Zhang W, Ni C, Gu Y, Hu J. J. Am. Chem. Soc. 2012; 134: 16999
- 13f Shen X, Miao W, Ni C, Hu J. Angew. Chem. Int. Ed. 2014; 53: 775
- 14 When the corresponding NH-sulfoximine was applied as starting material under the same conditions, no conversion was observed.
- 15 Notably, this behavior is in analogy to observations made in sulfoximine chemistry where the diastereomers of β-hydroxy sulfoximines stemming from ketone additions proved readily separable, while products from reactions with aldehydes were difficult to isolate in diastereomerically pure form. For a representative report, see: Johnson CR, Stark CJ. J. Org. Chem. 1982; 47: 1193
For recent examples, see:
For recent preparative approaches towards sulfondiimines, see:
For selected applications of sulfondiimines, see:
For selected syntheses and applications of structurally analogous NH-β-hydroxy sulfoximines that have been studied extensively, see: