

Subscribe to RSS
DOI: 10.1055/s-0045-1814731
Synchronous Bilateral Shamblin Type III Carotid Body Tumors and Adrenal Pheochromocytoma with SDHD Mutation: A Rare Presentation, with Multimodality Imaging Findings
Authors


Abstract
Pheochromocytomas and paragangliomas are relatively rare tumors, with an incidence of approximately 0.6 cases per 100,000 person-years. Moreover, the co-occurrence of these tumors is extremely rare and is often associated with pheochromocytomas/paragangliomas-related pathogenic mutations. We present the case of a 33-year-old female diagnosed with bilateral carotid body paragangliomas and a concurrently detected pheochromocytoma, with metastases to the abdominal lymph nodes and lung. DNA analysis revealed a mutation in the succinate dehydrogenase subunit D gene. The tumors displayed high-grade SSTR expression (Krenning grade 4 uptake) on Gallium-68 [⁶⁸Ga]-DOTA-(Tyr3)-octreotate Positron Emission Tomography/Computed Tomography, with no significant tracer concentration on the I-131 MIBG scan. Another notable feature in this case was the visually evident intra- and inter-tumoral metabolic heterogeneity on 18F-fluorodeoxyglucose positron emission tomography/computed tomography, especially within the multiple carotid paragangliomas. With the adoption of a multimodality diagnostic approach (MRI, FDG PET/CT, SSTR PET/CT, and I-131 MIBG scintigraphy), a holistic theranostic approach was employed with the most rational therapeutic option offered to the patient.
Keywords
carotid body tumors - PET/CT - 18F-FDG - 68Ga-DOTATATE - 131I-MIBG - pheochromocytoma - Ki-67 - histopathological analysisIntroduction
Paragangliomas and pheochromocytomas are neuroendocrine-origin neoplasms that may arise sporadically or in association with hereditary syndromes. Paragangliomas are either derived from the paravertebral sympathetic chains or parasympathetic ganglia in the body, whereas pheochromocytomas arise from chromaffin cells of the adrenal medulla.[1] [2]
Clinical manifestations may vary depending on the location and functionality of these tumors. Sympathetic paragangliomas and pheochromocytomas typically present with symptoms attributable to catecholamine excess, such as blood pressure fluctuations (episodic hypertension or orthostatic hypotension), palpitations, diaphoresis, and headache.[3] On the other hand, head and neck paragangliomas are usually non-functioning and more commonly discovered incidentally during imaging studies. When symptomatic, they present with compressive or infiltrative symptoms on adjacent structures, causing hearing loss, tinnitus, dysphagia, and even cranial nerve palsies.[1]
Anatomical and functional imaging play complementary roles in the diagnosis, staging, and treatment planning of pheochromocytomas/paragangliomas (PPGLs). Functional radionuclide imaging offers a range of radiotracers, such as Gallium-68 [68Ga]-DOTA-(Tyr3)-octreotate Positron Emission Tomography/Computed Tomography (68Ga-DOTATATE), 131I-metaiodobenzylguanidine (123I-MIBG), 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT), and 18F-Flourodopamine (18F-FDOPA), tailored to imagine the underlying molecular characteristics and pathogenic pathways of these neoplasms.[4]
Herein, we present a case of bilateral carotid body paragangliomas with synchronous retroperitoneal pheochromocytoma in a young female, associated with a succinate dehydrogenase subunit D (SDHD) mutation. The presentation is relatively rare, with only a few cases reported worldwide.[5] This report aims to highlight the importance of multimodality functional imaging and genetic analysis for the characterization and optimization of treatment strategies in such complex paraganglioma–pheochromocytoma syndromes.
Case Report
A 33-year-old-female presented with bilateral neck swellings that has been present for the past 3 years, with a rapid increase in size over the previous 3 to 4 weeks. The patient also reported a history of multiple episodes of postural hypotension and a single episode of accelerated hypertension. On biochemical evaluation, plasma normetanephrine levels were elevated (760 pg/mL).
Magnetic resonance imaging (MRI) of the neck revealed two well-defined lobulated soft-tissue masses located at the bilateral carotid bifurcations, measuring approximately 3.9 × 4 × 8.6 cm on the left side and 2.5 × 2.7 × 4.1 cm on the right side (AP × TR × CC). Characteristic splaying of the internal and external carotid arteries, with 100% vascular encasement, was noted bilaterally. A diagnosis of bilateral carotid paragangliomas (100% encasement of carotid arteries) was made based on clinical and imaging findings. Biopsy for histopathological analysis was intentionally deferred, considering the high risk of intraoperative bleeding and potential catecholamine crisis associated with functional paragangliomas, particularly in those with extensive vascular encasement.
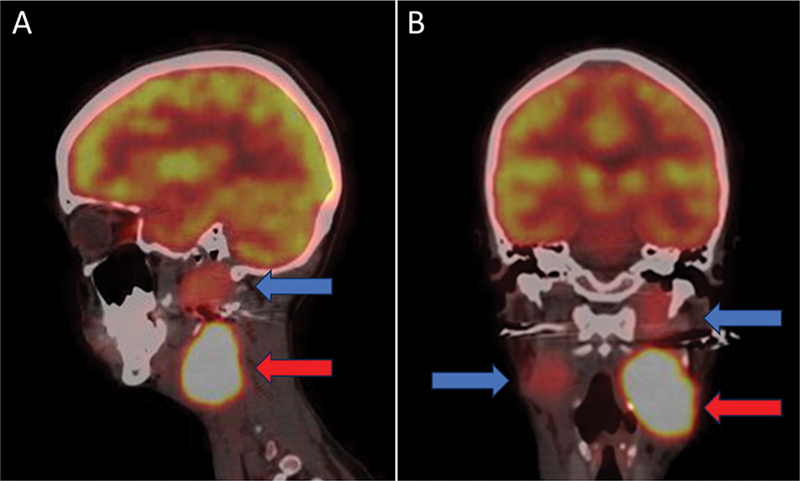
Diagnostic 68Ga-DOTATATE PET/CT was performed for disease characterization and staging, which revealed intense SSTR expression (Krenning grade 4) in the bilateral carotid paragangliomas. Additionally, an SSTR expressing soft-tissue lesion was noted in the retroperitoneal aortocaval region, measuring approximately 2 × 1.6 cm, without any evidence of locoregional invasion of adjacent structures. A few SSTR expressing locoregional retroperitoneal lymph nodes were also noted. Moreover, multiple SSTR-expressing soft-tissue nodules were scattered in the bilateral lung parenchyma ([Fig. 1]).

Excision of the retroperitoneal mass was done at a different center, and histopathological analysis was suggestive of pheochromocytoma, with a Ki-67 index of 2 to 3%. Genetic sequencing revealed a mutation in the succinate SDHD gene. Considering the extensive encasement of the bilateral carotid arteries leading to inoperability of the tumors, and extensive metastatic disease, the patient was referred for systemic therapy. 18F-FDG PET/CT scan was performed to further assess tumor biology, revealing heterogenous low-to-moderate grade tracer uptake in the bilateral carotid paragangliomas, left para-aortic lymph node, and few lung nodules ([Fig. 2]). 131I-MIBG scintigraphy performed for theranostic evaluation, revealed no significant radiotracer uptake in the bilateral carotid body tumors or metastatic lesions ([Fig. 3]).

Discussion
The diagnostic evaluation of PPGLs involves a multidisciplinary approach, including biochemical markers, anatomical and functional imaging, and genetic analysis. Biochemical assessment includes measurement of plasma-free metanephrines and normetanephrines, or urinary fractionated metanephrines, and is primarily used for screening purposes. Anatomical imaging using MRI or CT aids in the localization and delineation of the primary tumors, with MRI being preferred for head and neck paragangliomas due to its superior soft-tissue resolution and lack of radiation exposure.[6]
Functional imaging is essential for disease characterization, detection of small metastatic foci, and prognostication. 68Ga-DOTA-conjugated somatostatin analogues imaging has proven to be highly sensitive for the detection of sporadic PPGLs, especially in cases with SDHx-associated mutations.[7] This modality is particularly useful for the evaluation of lesions where histopathological analysis is not feasible, such as the bilateral carotid body tumors and multiple lung nodules in this patient.
On the other hand, 18F-FDG PET/CT is valuable for the assessment of metastatic or aggressive disease, especially in succinate dehydrogenase (SDH) subtype-B mutation carriers. 18F-FDOPA PET/CT imaging offers excellent sensitivity for imaging both hereditary and sporadic pheochromocytomas, in addition to imaging head and neck paragangliomas.[8] Although 123I-MIBG imaging holds high specificity for imaging adrenal pheochromocytomas, and has theranostic implications in patient selection for targeted 131I-MIBG therapy, its limited sensitivity may lead to underestimation of disease burden in many cases.[9]
Approximately 40% of PPGLs have a hereditary basis, involving multiple germline mutations and syndromic associations such as von Hippel–Lindau, multiple endocrine neoplasia type 2 (RET), neurofibromatosis type 1, and hereditary paraganglioma–pheochromocytoma syndrome.[10] Notably, pathogenic mutations in the SDH complex subunits (SDHA, SDHB, SDHC, SDHD) are strongly associated with aggressive hereditary PPGL syndromes. SDHD mutations exhibit a strong predilection for head and neck paragangliomas, with a higher risk of metastatic potential compared to sporadic cases.[11] These findings were in concordance with the histopathological findings in our case.
The management of bilateral carotid body tumors or multifocal PPGLs has not yet been standardized. The current mainstay of treatment for carotid body tumors involves staged excision, with adequate preoperative alpha- and beta-adrenergic blockade,[12] although this option is associated with a significant risk of hemorrhage and neurovascular complications (especially in Shamblin type III tumors).[13] Other therapeutic options for locally advanced, multifocal, or metastatic disease include observation, intensity-modulated radiotherapy, radionuclide therapy—including peptide receptor radionuclide therapy (PRRT) or MIBG therapy—chemotherapy, and/or novel targeted therapies (e.g., tyrosine kinase inhibitors).[14] [15] In our case, considering the intense SSTR expression and absence of MIBG avidity ([Fig. 3]), the patient was deemed unsuitable for MIBG-based therapy. She was therefore planned for PRRT with 177Lu-DOTATATE ([Fig. 4]). These findings highlight the importance of using complementary imaging modalities for accurate staging and treatment planning in PPGLs.

Conclusion
In summary, this case illustrates a rare presentation of SDHD-mutated hereditary paraganglioma–pheochromocytoma syndrome, presenting as bilateral Shamblin III carotid body paragangliomas, with synchronous retroperitoneal pheochromocytoma and distant metastatic lesions in a young female. This case report highlights the importance of comprehensive multimodality functional imaging to understand disease pathology, evaluate inter- and intra-tumoral heterogeneity, and subsequently select the best therapeutic options based on the degree of uptake in metastatic lesions. Malignancies such as neuroendocrine tumors and paragangliomas may display variable patterns of tracer uptake, guiding personalized treatment choices for different patients, facilitated by nuclear medicine and molecular imaging.
Conflict of Interest
None declared.
-
References
- 1 Neumann HPH, Young Jr WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med 2019; 381 (06) 552-565
- 2 Tzikos G, Menni A, Cheva A. et al. Composite paraganglioma of the celiac trunk: a case report and a comprehensive review of the literature. Front Surg 2022; 9: 824076
- 3 Aygun N, Uludag M. Pheochromocytoma and paraganglioma: from clinical findings to diagnosis. Sisli Etfal Hastan Tip Bul 2020; 54 (03) 271-280
- 4 Ilias I, Chen CC, Carrasquillo JA. et al. Comparison of 6 different PET tracers for the localization of pheochromocytoma. J Nucl Med 2008; 49 (10) 1613-1619
- 5 Han EJ, Lee SH, Song IU, Chung YA, Maeng LS. Simultaneous adrenal pheochromocytoma and carotid body paraganglioma in a woman. Nucl Med Mol Imaging 2013; 47 (01) 67-68
- 6 Thelen J, Bhatt AA. Multimodality imaging of paragangliomas of the head and neck. Insights Imaging 2019; 10 (01) 29
- 7 Janssen I, Chen CC, Taieb D. et al. 68Ga-DOTATATE PET/CT in the localization of head and neck paragangliomas compared to other functional imaging modalities and CT/MRI. J Nucl Med 2016; 57 (02) 186-191
- 8 Taïeb D, Timmers HJLM, Hindié E. et al; European Association of Nuclear Medicine. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2012; 39 (12) 1977-1995
- 9 Ilias I, Divgi C, Pacak K. Current role of metaiodobenzylguanidine in the diagnosis of pheochromocytoma and medullary thyroid cancer. Semin Nucl Med 2011; 41 (05) 364-368
- 10 Gupta G, Pacak K. AACE Adrenal Scientific Committee. Precision medicine: an update on genotype/biochemical phenotype relationships in pheochromocytoma/paraganglioma patients. Endocr Pract 2017; 23 (06) 690-704
- 11 Andrews KA, Ascher DB, Pires DEV. et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD . J Med Genet 2018; 55 (06) 384-394
- 12 Lenders JW, Duh QY, Eisenhofer G. et al; Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99 (06) 1915-1942
- 13 Robertson V, Poli F, Hobson B, Saratzis A, Ross Naylor A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur J Vasc Endovasc Surg 2019; 57 (04) 477-486
- 14 Basu S, Abhyankar A, Jatale P. The current place and indications of 131I-metaiodobenzylguanidine therapy in the era of peptide receptor radionuclide therapy: determinants to consider for evolving the best practice and envisioning a personalized approach. Nucl Med Commun 2015; 36 (01) 1-7
- 15 Brewczyński A, Kolasińska-Ćwikła A, Jabłońska B, Wyrwicz L. Pheochromocytomas and paragangliomas—current management. Cancers (Basel) 2025; 17 (06) 1029
Address for correspondence
Publication History
Article published online:
05 January 2026
© 2026. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
-
References
- 1 Neumann HPH, Young Jr WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med 2019; 381 (06) 552-565
- 2 Tzikos G, Menni A, Cheva A. et al. Composite paraganglioma of the celiac trunk: a case report and a comprehensive review of the literature. Front Surg 2022; 9: 824076
- 3 Aygun N, Uludag M. Pheochromocytoma and paraganglioma: from clinical findings to diagnosis. Sisli Etfal Hastan Tip Bul 2020; 54 (03) 271-280
- 4 Ilias I, Chen CC, Carrasquillo JA. et al. Comparison of 6 different PET tracers for the localization of pheochromocytoma. J Nucl Med 2008; 49 (10) 1613-1619
- 5 Han EJ, Lee SH, Song IU, Chung YA, Maeng LS. Simultaneous adrenal pheochromocytoma and carotid body paraganglioma in a woman. Nucl Med Mol Imaging 2013; 47 (01) 67-68
- 6 Thelen J, Bhatt AA. Multimodality imaging of paragangliomas of the head and neck. Insights Imaging 2019; 10 (01) 29
- 7 Janssen I, Chen CC, Taieb D. et al. 68Ga-DOTATATE PET/CT in the localization of head and neck paragangliomas compared to other functional imaging modalities and CT/MRI. J Nucl Med 2016; 57 (02) 186-191
- 8 Taïeb D, Timmers HJLM, Hindié E. et al; European Association of Nuclear Medicine. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2012; 39 (12) 1977-1995
- 9 Ilias I, Divgi C, Pacak K. Current role of metaiodobenzylguanidine in the diagnosis of pheochromocytoma and medullary thyroid cancer. Semin Nucl Med 2011; 41 (05) 364-368
- 10 Gupta G, Pacak K. AACE Adrenal Scientific Committee. Precision medicine: an update on genotype/biochemical phenotype relationships in pheochromocytoma/paraganglioma patients. Endocr Pract 2017; 23 (06) 690-704
- 11 Andrews KA, Ascher DB, Pires DEV. et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD . J Med Genet 2018; 55 (06) 384-394
- 12 Lenders JW, Duh QY, Eisenhofer G. et al; Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99 (06) 1915-1942
- 13 Robertson V, Poli F, Hobson B, Saratzis A, Ross Naylor A. A systematic review and meta-analysis of the presentation and surgical management of patients with carotid body tumours. Eur J Vasc Endovasc Surg 2019; 57 (04) 477-486
- 14 Basu S, Abhyankar A, Jatale P. The current place and indications of 131I-metaiodobenzylguanidine therapy in the era of peptide receptor radionuclide therapy: determinants to consider for evolving the best practice and envisioning a personalized approach. Nucl Med Commun 2015; 36 (01) 1-7
- 15 Brewczyński A, Kolasińska-Ćwikła A, Jabłońska B, Wyrwicz L. Pheochromocytomas and paragangliomas—current management. Cancers (Basel) 2025; 17 (06) 1029