

Subscribe to RSS
DOI: 10.1055/s-0045-1813710
Peculiarities of Neurofibromatosis–Noonan Syndrome: Clinical, Radiological, and Histological Characteristics Illustrated
Authors
Funding None.

Abstract
Neurofibromatosis–Noonan syndrome (NFNS) is an exceptional disease combining two autosomal dominant conditions: neurofibromatosis type 1 (NF1) and Noonan syndrome. Herein, we report the case of a 37-year-old female patient with a history of Hashimoto thyroiditis who exhibited typical symptoms of pheochromocytoma, which was confirmed with hormonal assessment. On physical examination, she exhibited cutaneous features of NF1. She also had short stature and dysmorphic syndrome consistent with NS. Thus, the diagnosis of NFNS was retained. A cerebral magnetic resonance imaging was carried out, revealing a temporal cavernoma. This case is, to our knowledge, the first report of NFNS with cerebral cavernoma, adding new insight into the clinical spectrum of this rare syndrome. Definitely, gaps remain in our understanding of NFNS, and therefore, other studies are warranted.
Keywords
neurofibromatosis–Noonan syndrome - pheochromocytoma - autoimmune thyroiditis - autoimmune diseases - central nervous system cavernous hemangiomaIntroduction
Neurofibromatosis–Noonan syndrome (NFNS) results from the combination of features from two autosomal dominant conditions: neurofibromatosis 1 (NF1) and Noonan syndrome (NS).
NF1 is a rather common genetic condition attributable to a germline mutation in the NF1 gene, affecting 1 in 3,000 births, with a broad spectrum of clinical manifestations dominated by skin, neurological, and skeletal aberrations.[1] The diagnosis is based on the updated criteria of the U.S. National Institutes of Health Consensus Development Conference.[2]
NS is also a frequent disease, with a prevalence of 1 to 2,500 individuals, resulting from mutations in the genes coding for components or regulators of the Ras/mitogen-activated protein kinase pathway, a pivotal element in cell differentiation, expansion, as well as its survival,[3] thus explaining its wide range of clinical manifestations. Nevertheless, short stature, distinctive craniofacial dysmorphia, and cardiovascular abnormalities represent the hallmark features of NS.[4] In addition to molecular testing, the diagnosis can be established via the Van der Burgt criterion.[5]
Herein, we report a unique case of NFNS with gripping comorbidities: Hashimoto thyroiditis, pheochromocytoma, and cerebral cavernoma, while reviewing the current literature on these occurrences.
Case Description
A 37-year-old woman with a family history of NF1 in her father and hypothyroidism in her paternal cousin. She had a notable personal history of Hashimoto thyroiditis and had regular menses since the age of 13 years. She was admitted to the intensive care department for severe hypoxemic infectious pneumonia. Upon her admission, she had severe paroxysmal hypertension and tachycardia associated with moderate hypokalemia of 3 mmol/L. She was treated with antibiotics, continuous positive airway pressure, and isosorbide dinitrate. She was then transferred to our Department of Endocrinology for suspicion of pheochromocytoma.
On physical examination, she was overweight, with a body mass index of 25.8 kg/m2. She had a short stature of 1.51 m, a goiter, and a blood pressure reading of 9/6 without orthostatic hypotension. She exhibited dysmorphic features associated with: low hairline ([Fig. 1]), relative macrocephaly, wide and long forehead, triangular facies, sparse eyebrows, hypertelorism with bilateral ptosis predominantly in the left eye, midface hypoplasia, and a short fourth metacarpal.
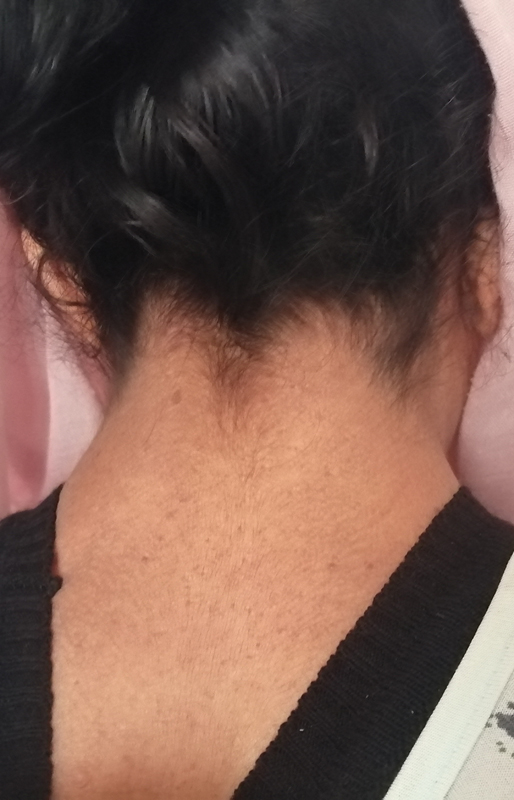
She also had axillary and facial freckles, disseminated cutaneous neurofibromas ([Fig. 2A, B]), seven café-au-lait macules exceeding 15 mm ([Fig. 2C]), and four Lisch nodules, thus making the diagnosis of NF1.

An ophthalmological examination revealed decreased bilateral visual acuity of 6/10, no papilledema, and a normal visual field. The neurological exam was normal. [Table 1] summarizes the clinical features of our patient.
Plasma free metanephrines were elevated: 61 times and 18 times the upper limit of normal for metanephrine and normetanephrine, respectively. Pelvic thoracic abdominal tomography showed a left heterogeneous adrenal tumor with central necrosis measuring 99 × 87 × 69 mm with a spontaneous density of 49 HU. Her transthoracic echography was normal.
Given the decreased visual acuity and the susceptibility of patients with NF1 to tumors, a cerebral magnetic resonance imaging was made, revealing a left occipitotemporal cavernoma of 25 × 26 mm ([Fig. 3]) and unidentified bright objects.

We initially started prazosin 5 mg/d; due to reflex tachycardia, a beta blocker was added, followed by intravenous hydration. A left adrenalectomy was carried out, and the postoperative period was uneventful. The anatomopathological exam demonstrated a pheochromocytoma, with a PASS score of 3 ([Fig. 4]). The patient was then lost to follow-up.

Discussion
Given the exceptional nature of NFNS, there are still no guidelines delineating the diagnostic modalities. However, the association of the clinical features of both conditions is the most reasonable approach.
The genetic background of NFNS is fertile ground for study. Many hypotheses have emerged to decipher its etiology. A recent systematic review of the literature demonstrated that the most plausible cause of NFNS is a mutation in the NF1 gene, with epigenetic factors contributing to the NS phenotype.[6]
As for clinical symptomatology, patients exhibit varying degrees of phenotypic characteristics of both diseases.[7] The index patient displayed typical signs of both NF1 and NS, and the diagnosis of NFNS was retained after consulting an experienced clinical geneticist. Unfortunately, genetic analysis was not available in our hospital.
Pheochromocytoma is a rare but well-studied entity that occurs in fewer than 5.7% of patients with NF1. As for sporadic cases, pheochromocytoma in NF1 patients occurs at a mean age of 42 years, is typically unilateral and nonmalignant.[8] In fact, inactivating mutations of NF1 result in the activation of PI3K/mToR leading to uncontrolled cellular proliferation and eventually tumorigenesis, such as pheochromocytoma.[9] To our knowledge, only one case of NFNS with pheochromocytoma has been reported in the literature by Jrad et al.[10]
Intriguingly, the index patient also had Hashimoto thyroiditis. The association between NFNS and autoimmune diseases (AID) was observed in only 4.3% of cases, according to a recent study.[6]
Many reports have emerged concerning the coexistence of AID and pheochromocytomas.[11] [12] [13] It seems that catecholamines alter the balance between Th1 helper lymphocytes and TH2 by activating β2 receptors. The predominance of TH2 results in AID.[12] Whether these associations are the result of chance or are attributable to a common pathophysiology is a matter of debate.
Our patient had another curious finding: a cerebral cavernoma. NS has been linked to cerebrovascular abnormalities such as cavernomas.[14] [15] In fact, activating mutations in RAF1, a gene implicated in NS[16] and encoding RAF kinase, have been implicated in endothelial cell development.[17] On the other hand, the association between NF1 and cerebral cavernoma is extremely rarely reported in the current literature.[18] Finally, we found no co-occurrence of NFNS and cerebral cavernoma in published studies. However, as explained above, association is biologically plausible.
Conclusion
NFNS is an extremely rare condition with an unknown genetic background that combines clinical features of two common genetic diseases, NF1 and NS. This case highlights the importance of comprehensive evaluation in NFNS patients, as rare comorbidities such as pheochromocytoma, autoimmune thyroiditis, and cerebral cavernoma may coexist and influence management. Significant gaps remain in our understanding of NFNS, and therefore, further studies are warranted.
Conflict of Interest
None declared.
Patient Consent Statement
The authors confirm that the patient provided written informed consent for publication.
Author Contribution
O.T. and R.H. contributed to the writing of the manuscript. E.H. was responsible for the conception of the study. B.B.A. and J.S. contributed to the study design. S.A. provided materials. S.H. was involved in data collection. H.M. performed the literature review. I.K. supervised the project. The data analysis was conducted by I.B. and A.Z.
Compliance with Ethical Principles
Formal ethical approval is not required for single cases and small case series.
-
References
- 1 Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers 2017; 3 (01) 17004
- 2 Choi J, An S, Lim SY. Current concepts of neurofibromatosis type 1: pathophysiology and treatment. Arch Craniofac Surg 2022; 23 (01) 6-16
- 3 Baldo F, Fachin A, Da Re B, Rubinato E, Bobbo M, Barbi E. New insights on Noonan syndrome's clinical phenotype: a single center retrospective study. BMC Pediatr 2022; 22 (01) 734
- 4 Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A. et al. [Noonan syndrome: genetic and clinical update and treatment options]. An Pediatr (Engl Ed) 2020; 93 (01) 61.e1-61.e14
- 5 Management of Noonan Syndrome - A Clinical Guideline: Noonan Syndrome Guideline Development Group. In: Noonan Syndrome [Internet]. Elsevier; 2019 [Jan 10, 2024]. p. 159–188. Accessed at: https://linkinghub.elsevier.com/retrieve/pii/B9780128153482099874
- 6 Trimeche O, Sakka R, Hajji E. et al. Portraying the full picture of neurofibromatosis-Noonan syndrome: a systematic review of literature. J Med Genet 2025; 62 (02) 109-116
- 7 Reig I, Boixeda P, Fleta B, Morenoc C, Gámez L, Truchuelo M. Neurofibromatosis-Noonan syndrome: case report and clinicopathogenic review of the Neurofibromatosis-Noonan syndrome and RAS-MAPK pathway. Dermatol Online J 2011; 17 (04) 4
- 8 Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol 1999; 162 (05) 1582-1586
- 9 Costa MHS, Ortiga-Carvalho TM, Violante AD, Vaisman M. Pheochromocytomas and paragangliomas: clinical and genetic approaches. Front Endocrinol (Lausanne) 2015; 6: 126
- 10 Jrad M, Ksentini M, Abid S. et al. Pheochromocytoma associated with neurofibromatosis type 1 and Noonan syndrome: a case report and literature review. Biomed J Sci Tech Res 2020; 31 (02) 24018-24024
- 11 Housni B, Elharroudi T, Soufi M, Bouziane M, Azzouzi A. Graves' disease allied with multiple pheochromocytoma. Indian J Endocrinol Metab 2013; 17 (02) 323-325
- 12 Marino G, Michielon A, Musumeci MB, Autore C. Takotsubo syndrome: hyperthyroidism, pheochromocytoma, or both? A case report. Eur Heart J Case Rep 2021; 5 (08) ytab270
- 13 Mnif F, Othmen WB, Ghorbel D, Elleuch M, Salah DB, Abid M. Pheochromocytoma/ganglioneuroma and auto-immunity: report of two cases. J Endocrinol Thyroid Res 2019; 4 (03) 1-5
- 14 Zarate YA, Lichty AW, Champion KJ, Clarkson LK, Holden KR, Matheus MG. Unique cerebrovascular anomalies in Noonan syndrome with RAF1 mutation. J Child Neurol 2014; 29 (08) NP13-NP17
- 15 Tanaka Y, Masuno M, Iwamoto H. et al. Noonan syndrome and cavernous hemangioma of the brain. Am J Med Genet 1999; 82 (03) 212-214
- 16 Kobayashi T, Aoki Y, Niihori T. et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat 2010; 31 (03) 284-294
- 17 Wimmer R, Cseh B, Maier B, Scherrer K, Baccarini M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-α complex. Dev Cell 2012; 22 (01) 158-171
- 18 Rerat K, Parker F, Nasser G. et al. Occurrence of multiple cerebral cavernous malformations in a patient with neurofibromatosis type 1. J Neurol Sci 2015; 350 (1–2): 98-100
Address for correspondence
Publication History
Article published online:
03 December 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Thieme Medical and Scientific Publishers Pvt. Ltd.
A-12, 2nd Floor, Sector 2, Noida-201301 UP, India
-
References
- 1 Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers 2017; 3 (01) 17004
- 2 Choi J, An S, Lim SY. Current concepts of neurofibromatosis type 1: pathophysiology and treatment. Arch Craniofac Surg 2022; 23 (01) 6-16
- 3 Baldo F, Fachin A, Da Re B, Rubinato E, Bobbo M, Barbi E. New insights on Noonan syndrome's clinical phenotype: a single center retrospective study. BMC Pediatr 2022; 22 (01) 734
- 4 Carcavilla A, Suárez-Ortega L, Rodríguez Sánchez A. et al. [Noonan syndrome: genetic and clinical update and treatment options]. An Pediatr (Engl Ed) 2020; 93 (01) 61.e1-61.e14
- 5 Management of Noonan Syndrome - A Clinical Guideline: Noonan Syndrome Guideline Development Group. In: Noonan Syndrome [Internet]. Elsevier; 2019 [Jan 10, 2024]. p. 159–188. Accessed at: https://linkinghub.elsevier.com/retrieve/pii/B9780128153482099874
- 6 Trimeche O, Sakka R, Hajji E. et al. Portraying the full picture of neurofibromatosis-Noonan syndrome: a systematic review of literature. J Med Genet 2025; 62 (02) 109-116
- 7 Reig I, Boixeda P, Fleta B, Morenoc C, Gámez L, Truchuelo M. Neurofibromatosis-Noonan syndrome: case report and clinicopathogenic review of the Neurofibromatosis-Noonan syndrome and RAS-MAPK pathway. Dermatol Online J 2011; 17 (04) 4
- 8 Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol 1999; 162 (05) 1582-1586
- 9 Costa MHS, Ortiga-Carvalho TM, Violante AD, Vaisman M. Pheochromocytomas and paragangliomas: clinical and genetic approaches. Front Endocrinol (Lausanne) 2015; 6: 126
- 10 Jrad M, Ksentini M, Abid S. et al. Pheochromocytoma associated with neurofibromatosis type 1 and Noonan syndrome: a case report and literature review. Biomed J Sci Tech Res 2020; 31 (02) 24018-24024
- 11 Housni B, Elharroudi T, Soufi M, Bouziane M, Azzouzi A. Graves' disease allied with multiple pheochromocytoma. Indian J Endocrinol Metab 2013; 17 (02) 323-325
- 12 Marino G, Michielon A, Musumeci MB, Autore C. Takotsubo syndrome: hyperthyroidism, pheochromocytoma, or both? A case report. Eur Heart J Case Rep 2021; 5 (08) ytab270
- 13 Mnif F, Othmen WB, Ghorbel D, Elleuch M, Salah DB, Abid M. Pheochromocytoma/ganglioneuroma and auto-immunity: report of two cases. J Endocrinol Thyroid Res 2019; 4 (03) 1-5
- 14 Zarate YA, Lichty AW, Champion KJ, Clarkson LK, Holden KR, Matheus MG. Unique cerebrovascular anomalies in Noonan syndrome with RAF1 mutation. J Child Neurol 2014; 29 (08) NP13-NP17
- 15 Tanaka Y, Masuno M, Iwamoto H. et al. Noonan syndrome and cavernous hemangioma of the brain. Am J Med Genet 1999; 82 (03) 212-214
- 16 Kobayashi T, Aoki Y, Niihori T. et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat 2010; 31 (03) 284-294
- 17 Wimmer R, Cseh B, Maier B, Scherrer K, Baccarini M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-α complex. Dev Cell 2012; 22 (01) 158-171
- 18 Rerat K, Parker F, Nasser G. et al. Occurrence of multiple cerebral cavernous malformations in a patient with neurofibromatosis type 1. J Neurol Sci 2015; 350 (1–2): 98-100