

Subscribe to RSS
DOI: 10.1055/s-0045-1811947
miR-1 Promotes Apoptosis and Aggravates Myocardial Ischemia–Reperfusion Injury by Downregulating Insulin-Like Growth Factor-1
Authors
Funding This study was supported by the National Natural Science Foundation of China (81473453, 81673800) and the Projects of International Science and Technology Cooperation in Henan (182102410084).

Abstract
Objective
MicroRNA-1 (miR-1) aggravates myocardial ischemia–reperfusion (I/R) injury, whereas insulin-like growth factor-1 (IGF-1) maintains cardiomyocyte homeostasis. In this study, the aim is to investigate whether miR-1 can exacerbate I/R injury through the regulation of IGF-1.
Methods
The infarct area, lactate dehydrogenase, miR-1 level, and apoptosis level were examined in the Langendorff isolated rat I/R model. The hypoxia–reoxygenation model of rat cardiac myocytes and H9c2 cells were developed to determine the levels of miR-1, IGF-1 mRNA, and IGF-1 protein. Furthermore, the dual-luciferase assay was used to verify the relationship between miR-1 and IGF-1.
Results
Overexpression of miR-1 increased the level of apoptosis and decreased the IGF-1 expression. However, inhibition of miR-1 expression decreased the level of apoptosis, alleviated the degree of injury, and increased the IGF-1 expression. Overexpression of IGF-1 also reduced the degree of cellular damage and level of apoptosis caused by the overexpression of miR-1. When IGF-1 was knocked down, myocardial cells displayed more severe damage and a higher apoptosis level, even with decreased levels of miR-1.
Conclusion
miR-1 promotes apoptosis and aggravates I/R injury by downregulating IGF-1.
Keywords
miR-1 - insulin-like growth factor-1 - myocardial ischemia–reperfusion injury - hypoxia–reperfusion injury - apoptosisIntroduction
Acute myocardial infarction (AMI) is one of the leading causes of cardiovascular morbidity and mortality. The widespread application of AMI treatment methods, such as thrombolysis, interventional therapy, and coronary artery bypass grafting, has played an important role in the survival of dying myocardium or the reduction of myocardial infarction area. However, since myocardial ischemia–reperfusion (I/R) injury has become increasingly prominent, it has attracted a growing amount of attention.[1] The main pathological basis of I/R is cardiomyocyte apoptosis, with the level of apoptosis determining the degree of I/R injury and clinical prognosis.[2]
MicroRNA-1 (miR-1) is a type of miRNA that is abundant in cardiomyocytes and is directly involved in the occurrence and development of heart disease. Previous studies have identified miR-1 as a marker of cardiac stress; its expression is upregulated during I/R injury and is increased in the peripheral blood of patients with AMI. In a mouse myocardial ischemia model, the level of miR-1 was higher than that in the control group. We previously discovered that inhibiting miR-1 expression reduced the apoptosis of H9c2 cardiomyocytes induced by H2O2 in rats.[3] Conversely, increased miR-1 expression promoted cardiomyocyte apoptosis by increasing the expression of cytochrome C and Caspase apoptotic proteins.[4] Myocardial I/R injury is classified as “chest impediment” or “true heart pain” in traditional Chinese medicine (TCM). “qi deficiency and blood stasis” is core pathogenesis. Modern pharmacological research on Chinese medicine indicates that blood-activating and stasis-resolving herbs, such as honghua (safflower), can ameliorate myocardial injury by modulating the expression of miRNA-1. Although miR-1 promotes the apoptosis of cardiomyocytes, its mechanism remains unknown.
Insulin-like growth factor-1 (IGF-1) is an important regulator of cell homeostasis.[5] In hypoxic myocardium, the expression of IGF-1 is decreased, while apoptosis of cardiomyocytes is increased.[6] In addition, the knockout of IGF-1 promotes apoptosis. However, high IGF-1 expression contributes to cell proliferation and reduces the degree of hypoxia-induced damage.[7] Studies found that IGF-1 is a target gene for several miRNAs. For instance, miR-320 regulates cardiomyocyte apoptosis and I/R through targeting IGF-1.[8] According to the prediction of the Targestscan website, there are multiple miR-1 binding sites in the 3′UTR region of IGF-1 mRNA. Therefore, we hypothesize that miR-1 may play a role in promoting cardiomyocyte apoptosis and aggravating I/R injury by targeting IGF-1 during the I/R injury process.
In this study, an isolated rat I/R model and adult rat primary cardiomyocyte or H9c2 cardiomyocyte hypoxia–reoxygenation (H/R) model were used to determine the expression of miR-1 in myocardial tissue and cells, as well as the effects of miR-1 on the myocardial infarction area and apoptosis. Furthermore, the interaction between miR-1 and IGF-1 was verified by enhancing or inhibiting miR-1 and IGF-1 expression, as well as using the dual luciferase assay. This study reveals the role of miR-1 in aggravating I/R injury and provides a novel therapeutic strategy for protecting cardiomyocytes under AMI-like cardiovascular diseases.
Materials
Experimental Animals
Healthy SPF male SD rats, weight (230 ± 20) g, were purchased from the Henan Experimental Animal Center. The animal certificate number was number: 41003100006612. All animal experiments were approved by the Ethics Committee of Henan Province Hospital of Traditional Chinese Medicine (The Second Affiliated Hospital of Henan University of Chinese Medicine), Ethics Approval Number: PZ-HNSZYY-2019-072. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Reagents
Fetal bovine serum (FBS, Biological Industries, Beit-Haemek, Israel, Cat# 04-001-1ACS). Albumin Bovine, Collagenase 2-TYPE II Gibco (Solarbio, Beijing, China, Cat# A8020, 17101015). Lactate dehydrogenase (LDH) kit, Micro BCA protein quantification kit (Beyotime Biotechnology, Shanghai, China, Cat# C0017, AR1110). SDS-PAGE gel preparation kit (Genshare, Xi'an, China, Cat# JC-PE022). miRNA first-strand cDNA synthesis (stem-loop method) (Sangon BioTech, Shanghai, China, Cat# B532453). RT kit with gDNA eraser (Gene-Better Biotechnology, Beijing, China, Cat# P514-100). Anti-BAX antibody, anti-Bcl-2 antibody (Propeintech, Wuhan, China, Cat# 50599-2, 26593-1). Anti-IGF-1 antibody (Abcam, London, England, Cat# ab53154). Joklik (Sigma, St. Louis, Missouri, United States, Cat# M0518-10L).
Instruments
Western blotting system, gel imaging system, electrophoresis instrument, and electrophoresis tank (Bio-rad, United States). Thermo Forma 3111 carbon dioxide incubator (Thermo Fisher Scientific, United States). ABI 7500 Real-time Quantitative PCR (qPCR) System (Applied Biosystems, United States). BD FACS Canto II flow cytometer (BD, United States). Glomax 20 dual-luciferase detector (Promega, United States).
Methods
Isolated Rat Heart Perfusion
The cardiac I/R model was established according to Chen et al.[9] The heart was quickly removed and placed in precooled KH buffer (118 mmol/L NaCl, 4.75 mmol/L KCl, 1.1 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 25 mmol/L NaHCO3, 1.4 mmol/L CaCl2, 7 mmol/L D-glucose, 1.4 mmol/L bovine serum albumin [BSA]). Then, the heart was fixed and perfused. The left ventricular systolic pressure and left ventricular end-diastolic pressure was measured by a balloon catheter. The model group was perfused for 30 minutes, paused for 40 minutes, then perfused again for 1 hour. The control group was continuously perfused for 130 minutes. Next, the tissues were quickly frozen and stored at −80°C.
Isolation and Culture of Adult Rat Primary Cardiomyocytes
Rat cardiomyocytes were isolated according to Qi et al.[10] The heart was quickly removed and placed in precooled Joklik solution (11.23 g Joklik, 2 g NaHCO3, 0.144 g MgSO4, and 0.198 g L-Carnitine were diluted to 1 L with water). The perfusion lasted for 30 to 45 minutes with collagenase and was terminated after the heart had softened. The solution was then filtered through a 100-mesh filter and centrifuged at 2,000 r/min for 20 seconds. Following that, the cells were resuspended in M199+ culture medium (1.19 g HEPES, 0.04 g L-Carnitine, 2 mL penicillin–streptomycin, 2 g BSA, and M199 were diluted to 200 mL).
The culture medium was subsequently replaced with Dulbecco's modified Eagle medium (DMEM) without sugar and serum. The cells were cultured at 37°C for 3 hours in an anoxic chamber with 95% N2 and 5% CO2. The medium was then replaced with M199 + , and the cells were cultured for 30 minutes before being collected for analysis.
H9c2 Cardiomyocyte Culture
H9c2 cells were purchased from the Shanghai Cell Resource Center of Chinese Academy of Sciences (lot number: GNR 5) and cultured in high-glucose DMEM containing 10% FBS.
The cells were seeded into 6-well plates at a density of 1.2 × 105 cells/well, cultured until the fusion degree was 65 to 75%, then starved overnight. After the medium was replaced with low-sugar DMEM, the cells were cultured at 37°C for 6 hours in an anoxic chamber with 95% N2 and 5% CO2. The medium was then replaced with high-sugar DMEM with 10% FBS. The cells were cultured for a further 24 hours before being collected for analysis.
Lactate Dehydrogenase Release Detection
Each well was added 120 μL of perfusion solution or cell supernatant, followed by the addition of 60 μL LDH detection solution. After mixing and incubating at room temperature in the dark for 30 minutes, the absorbance was measured at 490 nm.
Detection of Myocardial Infarction Area
The rat hearts were frozen at −20°C for 30 minutes, cut into 2-mm-thick tissues, then incubated with 2% TTC solution at 37°C in the dark for 30 minutes. Following that, the heart slices were fixed with formaldehyde for 6 hours and photographed. The total area and infarct area of the heart slices were determined using Image J. The infarct rate of myocardial tissue was calculated based on the ratio of infarct area to total area.
Detection of Myocardial Apoptosis Rate
The rat hearts were fixed with formaldehyde, embedded in paraffin, and sequentially incubated with protease K working solution, DNase I buffer, fluorescein isothiocyanate (FITC)-12-dUTP staining solution, and DAPI staining solution. The observation was conducted at 520 and 460 nm. The apoptosis rate was determined according to the ratio of green fluorescence number to blue fluorescence number.
Cell Transfection and Lentivirus Infection
In order to transfect H9c2 cells, Lipofectamine 2000 (Invitrogen) was mixed with the miR-1-3p inhibitor, NC inhibitor, IGF-1-siRNA, siRNA-NC, or miR-1-3p inhibitor + IGF-1-siRNA. After 48 hours of transfection, the transfection efficiency was determined by qRT-PCR. The cells were preintervened as required before being collected for analysis.
The lentivirus for IGF-1 overexpression (Lenti-IGF-1) was used to infect the H9c2 cells. Lenti-GFP was used as the negative control. After a 48-hour infection period, the cells were collected for analysis.
Real-Time Quantitative Polymerase Chain Reaction
Total RNA was extracted using the Trizol kit, and miRNAs were extracted using the miRNA kit, then reverse transcribed into cDNA using the reverse transcription kit. miR-1 was reversely transcribed using the neck-loop method. The SYBR Green PCR Master Mix was used for RNA quantification of IGF-1, Bcl-2, Bax mRNA, and miR-1. Reaction system (20 μL): SYBR Premix Ex Taq 10 μL, each of the forward and reverse primers 0.5 μL, cDNA template 2 μL, ddH2O 6.4 μL. Reaction conditions: 95°C 30 seconds, 95°C 10 seconds, 60°C 30 seconds, 40 cycles. The annealing temperature was set at 60°C, and 40 cycles of amplification were selected. Following that, the 2−△△Ct method was used for analysis based on the Ct values. The primer sequences are shown in [Table 1].
Western Blotting
The total protein was extracted using RIPA lysate, and the protein concentration was determined by BCA. The total protein was then separated by SDS-PAGE electrophoresis and transferred onto a membrane. The membrane was blocked with 5% skimmed milk at room temperature for 1 hour at room temperature, incubated with anti-IGF-1, anti-Bcl-2, anti-Bax, and anti-GAPDH antibodies at 4°C overnight, dilution of primary antibody: IGF-1 (1:1000), Bcl-2 (1:1000), Bax (1:2000), GAPDH (1:3000), followed by incubation with a secondary antibody for 1 hour. The membrane was then developed with an ECL luminescent solution. The images were imaged using the Bio-Rad ChemiDoc XRS + Imaging System, and the target band was quantitatively analyzed.
Dual-Luciferase Reporter Assay
The dual-luciferase reporter assay was employed to validate the direct binding interaction between miR-1 and the 3′UTR of IGF-1. The targeted regulatory mechanism of miR-1 on IGF-1 was confirmed by comparing luminescence levels of wild-type and mutant-type reporter plasmids. Wild-type or mutant pmirGLO IGF-1 luciferase reporter plasmids, as well as miR-1-3p simulant or NC control, were cotransfected into HEK 293 cells. After 48 hours, the luciferase activity of the transfected cells was measured using the dual-luciferase reporter assay. The luciferase activity was determined based on the ratio of firefly to Renilla fluorescence activity.
Apoptosis Detected by Flow Cytometry
H9c2 cells were incubated with FITC-labeled annexin V and propidium iodide solution in the dark for 15 minutes. Data were obtained using the BD Jazz flow cytometer and analyzed using the FlowJO_10.0 software.
Statistical Analysis
All data were expressed as mean ± SD. Comparison among groups was conducted using the one-way analysis of variance or the t-test. p < 0.05 was considered statistically significant.
Results
Upregulation of miR-1 in the Ischemia–Reperfusion Rat Model and a Hypoxia–Reoxygenation Cell Model
After reperfusion, the left ventricular pressure difference (+dP/dt, maximum rate of the arterial pressure increase during systole) and heart rate significantly decreased, whereas −dP/dt (maximum rate of diastolic arterial pressure rise) increased in the I/R injury model ([Fig. 1A]). Furthermore, the myocardial infarction area was enlarged, and LDH content in the perfusion fluid (p < 0.05, [Fig. 1B, C]) and miR-1 expression (p < 0.05, [Fig. 1D]) significantly increased.

The primary myocardial cells of the adult rats were used to establish H/R injury model. The results demonstrated that the shape of the cells was significantly shortened ([Fig. 1E]), and LDH content in the supernatant significantly increased (p < 0.05, [Fig. 1F]). Similar results were also observed in the H9c2 cardiomyocyte H/R injury model ([Fig. 1H]), with significantly increased miR-1 (p < 0.05, [Fig. 1G, I]).
Cardiomyocyte Apoptosis Increases in Both the Ischemia–Reperfusion Rat Model and Hypoxia–Reoxygenation Cell Model
The number of apoptotic cells stained with TUNEL in the I/R injury model group was significantly higher than that in the control group (p < 0.05, [Fig. 2A]). The qPCR and Western blot results showed that there was an increase in Bax/Bcl-2 ratio (p < 0.05, [Fig. 2B, C]). In order to better understand the level of apoptosis, we established H/R injury models using isolated primary cardiomyocytes from adult rats and H9c2 cardiomyocytes. It showed a similar apoptosis occurrence in the above-mentioned I/R injury models ([Fig. 2D–H]).

Cardiomyocyte Apoptosis is Associated with Expression of miR-1 .
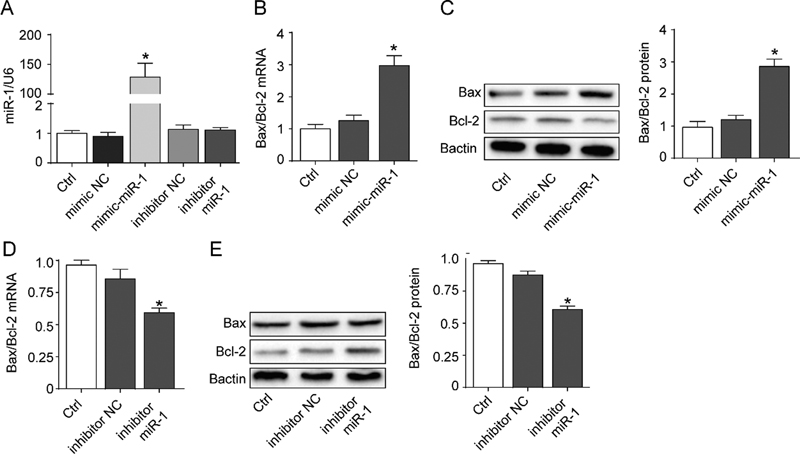
In order to study the relationship between miR-1 and apoptosis, H9c2 cell models with high or low miR-1 expression were developed ([Fig. 3A]). There was an increase in the ratio of Bax/Bcl-2 mRNA to protein when miR-1 mimic was applied (p < 0.05, [Fig. 3B, C]). Although transfection of miR-1 inhibitor did not significantly affect miR-1 expression, there was decrease in the ratio of Bax/Bcl-2 mRNA to protein (p < 0.05, [Fig. 3D, E]). These findings indicated that miR-1 expression positively correlated with apoptosis.
Insulin-Like Growth Factor-1 Inhibits Cardiomyocyte Apoptosis
The expression of IGF-1 mRNA and protein in the I/R injury model group was lower than that in the control group (p < 0.05, [Fig. 4A, B]). In order to better understand the relationship among IGF-1, cardiomyocyte apoptosis, and miR-1, we overexpressed IGF-1 in H9c2 cells. The results demonstrated that Lenti-IGF-1 decreased the rate of apoptotic cells, as well as the mRNA and protein ratio of Bax to Bcl-2 (p < 0.05, [Fig. 4C–E]). However, there was no change in the level of miR-1 (p > 0.05, [Fig. 4F]), indicating that IGF-1 regulates cardiomyocyte apoptosis without affecting miR-1 expression.

miR-1 Contributes to Cardiomyocyte Apoptosis by Inhibiting the Expression of Insulin-Like Growth Factor-1
In the H/R model of H9c2 cardiomyocytes, the apoptosis rate was increased and the expression of IGF-1 was decreased in the cotransfection group of miR-1 inhibitor and siIGF-1 (p < 0.05, [Fig. 5A]). Compared with the model group, the mRNA and protein ratio of Bax to Bcl-2 was increased, and the cotransfection of miR-1 inhibitor did not reverse the antiapoptotic effect of IGF-1 ([Fig. 5B–F]). On the other hand, when miR-1 mimic was cotransfected with lenti-IGF-1, the apoptosis rate was decreased ([Fig. 5G]). After overexpression of IGF-1, the mRNA and protein ratio of Bax to Bcl-2 was decreased. Similarly, miR-1 mimics did not exert a reversal effect ([Fig. 5H–L]).

Verification of Insulin-Like Growth Factor-1 as a Target Gene of miR-1
The binding and mutation sites in the 3′UTR of IGF-1 mRNA according to the miRNA-Target bioinformatics analysis are shown in [Fig. 6A]. The fluorescence activity of the IGF-1 wild-type dual-luciferase reporter gene in H9c2 cells in the miR-1 group significantly decreased compared with that in the NC group (p < 0.05, [Fig. 6B]), based on the dual-luciferase reporter gene experiment. The expression levels of IGF-1 mRNA and protein in the miR-1 inhibitor group significantly increased compared with those in the H/R model and NC model groups (p < 0.05, [Fig. 6C, D]). These findings suggested that miR-1 regulates IGF-1 by binding it directly.

Discussion
In this study, the levels of miR-1 and apoptosis significantly increased, while the level of IGF-1 decreased in both I/R injury myocardial tissues and H/R injury cardiomyocytes. Furthermore, the application of miR-1 inhibitor decreased the level of miR-1. In addition, the level of apoptosis significantly decreased, whereas the level of IGF-1 significantly increased. However, opposite effects were observed when miR-1 was overexpressed. The luciferase method validated that miR-1 directly bound to the 3′UTR region of IGF-1 and negatively regulated its expression. Therefore, miR-1 was able to regulate the degree of apoptosis and injury in cardiomyocytes. The miR-1/IGF-1 regulatory mechanism discovered in this study, whereby hydroxysafflor yellow A reduces myocardial cell damage by downregulating miR-1, aligns with the TCM principle of “activating blood circulation and resolving stasis” in treating I/R injury.[11] The above results demonstrated a promising strategy for the clinical development of new drugs to treat I/R injury.
Apoptosis is an active process that is directly regulated by many genes, including the Bcl-2 family, which is an important regulatory factor in the apoptosis process.[12] The Bcl-2 family is classified into antiapoptotic proteins, such as Bcl-2 and BCl-xL, and proapoptotic proteins, such as Bax and Bak.[13] During I/R injury, there is a decrease in mitochondrial membrane potential, flowout of cytochrome C, as well as the triggering and activation of downstream caspases that result in apoptosis.[14] Our results revealed that the apoptosis rate, as well as the ratio of Bax to Bcl-2 mRNA and protein in both the myocardial tissue I/R and cardiomyocyte H/R models, was significantly higher than that in the control group, suggesting that the level of apoptosis could effectively reflect the degree of I/R injury.
The expression of miR-1 was up-regulated in AMI and I/R injury.[15] [16] Furthermore, there was significantly increased miR-1 expression in the plasma of clinical MI patients.[17] miR-1 overexpression inhibits the viability of neonatal rat cardiomyocytes,[18] whereas inhibition of miR-1 reduces cardiomyocyte apoptosis and the myocardial infarction area.[19] The findings revealed that miR-1 positively regulates cardiomyocyte apoptosis. Our results also suggested that the expression of miR-1 increased in both I/R and H/R injuries. When miR-1 mimics were transfected in H9c2 cardiomyocytes, the ratio of Bax to Bcl-2 mRNA and protein increased. However, transfection with a miR-1 inhibitor decreased the level of apoptosis, which was consistent with previous reports. The above results indicated that miR-1 regulates cardiomyocyte apoptosis.
IGF-1 is a cytoprotective molecule that is used for I/R treatment.[20] [21] Previous studies have reported that the IGF-1 pathway protects cardiomyocytes from injury and promotes the differentiation of adipose stem cells into cardiomyocytes.[22] [23] Treatment with IGF-1 protects the heart and reduces apoptosis caused by I/R injury.[24] This study demonstrated that the expression of IGF-1 decreased in I/R injured tissue and the level of apoptosis increased in the damaged myocardium. Furthermore, the results confirmed that overexpression of IGF-1 significantly inhibited the apoptotic rate of H9c2 cardiomyocytes and decreased the ratio of Bax to Bcl-2 mRNA and protein, which was consistent with the previous reports.[25]
There is growing evidence that modulating the level of miRNAs to regulate cardiomyocyte apoptosis is becoming a new strategy for cardiovascular therapy. Both miR-185 and miR-29 regulated IGF-1, inhibited apoptosis caused by cardiomyocyte I/R injury and reduced the degree of injury.[25] [26] The downregulation of miR-320 inhibited cardiomyocyte apoptosis and reduced myocardial I/R injury by targeting IGF-1.[8] According to the prediction of the Targestscan website, there are multiple miR-1 binding sites in the 3 'UTR of IGF-1 mRNA. It was previously reported that miR-1 in smooth muscle cells regulates cell growth by targeting IGF-1.[27] In this study, the luciferase experiment validated that miR-1 directly binds to IGF-1 and negatively regulated its level. When a miR-1 inhibitor was transfected, the expression of IGF-1 mRNA and protein increased, whereas the level of apoptosis decreased. Simultaneous operation of transfection with a miR-1 inhibitor and silencing IGF-1 resulted in a significantly increased level of apoptosis. On the other hand, simultaneous operation of transfection with miR-1 mimics and overexpression of IGF-1 led to a significantly decreased level of apoptosis, validating that miR-1 regulates apoptosis through IGF-1. Finally, the results of the dual-luciferase assay suggested that miR-1 directly binds to IGF-1 to promote apoptosis.
Conclusion
In sum, increased miR-1 promotes apoptosis and aggravates the degree of I/R injury, whereas inhibiting miR-1 leads to myocardial tissue and cell protection. This effect is related to the negative regulation of IGF-1 expression and subsequent regulation of apoptosis, providing a potential clinical therapy for protecting cardiomyocytes under AMI-like cardiovascular diseases.
Conflict of Interest
The authors declare no conflict of interest.
CRediT Authorship' Contributions Statement
Zhen Lei and Fei Yan: Methodology, data curation, and writing -original draft. Yan Shu, Tengyun Liang, Mengwen Zhang, Xinzhou Wang, and Haixia Gao: Methodology, and formal analysis. Hong Wu: Methodology, project administration,and writing-review & editing.
-
References
- 1 Abo-Aly M, George B, Shokri E. et al. Cangrelor in addition to standard therapy reduces cardiac damage and inflammatory markers in patients with ST-segment elevation myocardial infarction. J Thromb Thrombolysis 2021; 52 (03) 934-940
- 2 Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020; 23 (03) 299-314
- 3 Hong W, Zhen L, Gao S. et al. Hydroxysafflower yellow pigment A protects against oxidative damage in H9c2 cardiomyocytes by inhibiting the expression of microRNA-1. J Zhejiang Chin Med Univ 2018; 41 (08) 636-641
- 4 Han Y, Cai Y, Lai X. et al. LncRNA RMRP prevents mitochondrial dysfunction and cardiomyocyte apoptosis via the miR-1–5p/hsp70 axis in LPS-induced sepsis mice. Inflammation 2020; 43 (02) 605-618
- 5 Liu T, Li F, Fei Y. et al. Serum insulin-like growth factor-1 as a potential prognostic biomarker for heart failure with reduced ejection fraction: a meta-analysis. Front Cardiovasc Med 2024; 11: 1415238
- 6 Lin M, Liu X, Zheng H. et al. IGF-1 enhances BMSC viability, migration, and anti-apoptosis in myocardial infarction via secreted frizzled-related protein 2 pathway. Stem Cell Res Ther 2020; 11 (01) 22
- 7 Baltazar-Lara R, Ávila-Mendoza J, Martínez-Moreno CG. et al. Neuroprotective effects of growth hormone (GH) and insulin-like growth factor type 1 (IGF-1) after hypoxic-ischemic injury in chicken cerebellar cell cultures. Int J Mol Sci 2020; 22 (01) 256
- 8 Song CL, Liu B, Diao HY. et al. Down-regulation of microRNA-320 suppresses cardiomyocyte apoptosis and protects against myocardial ischemia and reperfusion injury by targeting IGF-1. Oncotarget 2016; 7 (26) 39740-39757
- 9 Chen Y, Liu F, Chen BD. et al. rAAV9-mediated MEK1 gene expression restores post-conditioning protection against ischemia injury in hypertrophic myocardium. Cardiovasc Drugs Ther 2020; 34 (01) 3-14
- 10 Qi D, Atsina K, Qu L. et al. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest 2014; 124 (08) 3540-3550
- 11 Zhang C, Zhu HY, Wang J. et al. The mechanism of the inflammatory response of TCM in the prevention and treatment of cerebral ischemia-reperfusion injury based on the theory of blood circulation and blood stasis. J Zhejiang Chin Med Univ 2020; 44 (01) 107-110
- 12 Callens M, Kraskovskaya N, Derevtsova K. et al. The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer's disease. Biochim Biophys Acta Mol Cell Res 2021; 1868 (06) 118997
- 13 Lamb HM. Double agents of cell death: novel emerging functions of apoptotic regulators. FEBS J 2020; 287 (13) 2647-2663
- 14 Korshunova AY, Blagonravov ML, Neborak EV. et al. BCL2–regulated apoptotic process in myocardial ischemia–reperfusion injury (review). Int J Mol Med 2021; 47 (01) 23-36
- 15 Zhu X, Wang K, Jin Y. et al. Multiplexed fluorometric determination for three microRNAs in acute myocardial infarction by using duplex-specific nuclease and MoS2 nanosheets. Mikrochim Acta 2019; 187 (01) 15
- 16 Zhu WS, Guo W, Zhu JN. et al. Hsp90aa1: a novel target gene of miR-1 in cardiac ischemia/reperfusion injury. Sci Rep 2016; 6: 24498
- 17 Hao YL, Fang HC, Zhao HL. et al. The role of microRNA-1 targeting of MAPK3 in myocardial ischemia-reperfusion injury in rats undergoing sevoflurane preconditioning via the PI3K/Akt pathway. Am J Physiol Cell Physiol 2018; 315 (03) C380-C388
- 18 Gan J, Tang FMK, Su X. et al. microRNA-1 inhibits cardiomyocyte proliferation in mouse neonatal hearts by repressing CCND1 expression. Ann Transl Med 2019; 7 (18) 455
- 19 Hong T, Wei Y, Xue X. et al. A novel anti-coagulative nano complex in delivering miRNA-1 inhibitor against microvascular obstruction of myocardial infarction. Adv Healthc Mater 2020; 9 (11) e1901783
- 20 Tao L, Jia L, Li Y, Song C, Chen Z. Recent advances of adapter proteins in the regulation of heart diseases. Heart Fail Rev 2017; 22 (01) 99-107
- 21 Heinen A, Nederlof R, Panjwani P. et al. IGF1 treatment improves cardiac remodeling after infarction by targeting myeloid cells. Mol Ther 2019; 27 (01) 46-58
- 22 Qiao Y, Zhao Y, Liu Y. et al. miR-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun 2016; 477 (04) 541-547
- 23 Wang C, Liu W, Zhang X, Wang Y, Liu H, Li H. MEK/ERK signaling is involved in the role of VEGF and IGF1 in cardiomyocyte differentiation of mouse adipose tissue-derived stromal cells. Int J Cardiol 2017; 228: 427-434
- 24 Wang P, Gao R, Wu T. et al. Accumulation of endogenous adenosine improves cardiomyocyte metabolism via epigenetic reprogramming in an ischemia-reperfusion model. Redox Biol 2023; 67: 102884
- 25 Wang L, Niu X, Hu J. et al. After myocardial ischemia-reperfusion, miR-29a, and Let7 could affect apoptosis through regulating IGF-1. BioMed Res Int 2015; 2015: 245412
- 26 Wang R, Bao H, Zhang S, Li R, Chen L, Zhu Y. miR-186–5p promotes apoptosis by targeting IGF-1 in SH-SY5Y OGD/R model. Int J Biol Sci 2018; 14 (13) 1791-1799
- 27 Liu K, Ying Z, Qi X, Shi Y, Tang Q. MicroRNA-1 regulates the proliferation of vascular smooth muscle cells by targeting insulin-like growth factor 1. Int J Mol Med 2015; 36 (03) 817-824
Address for correspondence
Publication History
Received: 12 April 2025
Accepted: 17 July 2025
Article published online:
30 September 2025
© 2025. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Abo-Aly M, George B, Shokri E. et al. Cangrelor in addition to standard therapy reduces cardiac damage and inflammatory markers in patients with ST-segment elevation myocardial infarction. J Thromb Thrombolysis 2021; 52 (03) 934-940
- 2 Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020; 23 (03) 299-314
- 3 Hong W, Zhen L, Gao S. et al. Hydroxysafflower yellow pigment A protects against oxidative damage in H9c2 cardiomyocytes by inhibiting the expression of microRNA-1. J Zhejiang Chin Med Univ 2018; 41 (08) 636-641
- 4 Han Y, Cai Y, Lai X. et al. LncRNA RMRP prevents mitochondrial dysfunction and cardiomyocyte apoptosis via the miR-1–5p/hsp70 axis in LPS-induced sepsis mice. Inflammation 2020; 43 (02) 605-618
- 5 Liu T, Li F, Fei Y. et al. Serum insulin-like growth factor-1 as a potential prognostic biomarker for heart failure with reduced ejection fraction: a meta-analysis. Front Cardiovasc Med 2024; 11: 1415238
- 6 Lin M, Liu X, Zheng H. et al. IGF-1 enhances BMSC viability, migration, and anti-apoptosis in myocardial infarction via secreted frizzled-related protein 2 pathway. Stem Cell Res Ther 2020; 11 (01) 22
- 7 Baltazar-Lara R, Ávila-Mendoza J, Martínez-Moreno CG. et al. Neuroprotective effects of growth hormone (GH) and insulin-like growth factor type 1 (IGF-1) after hypoxic-ischemic injury in chicken cerebellar cell cultures. Int J Mol Sci 2020; 22 (01) 256
- 8 Song CL, Liu B, Diao HY. et al. Down-regulation of microRNA-320 suppresses cardiomyocyte apoptosis and protects against myocardial ischemia and reperfusion injury by targeting IGF-1. Oncotarget 2016; 7 (26) 39740-39757
- 9 Chen Y, Liu F, Chen BD. et al. rAAV9-mediated MEK1 gene expression restores post-conditioning protection against ischemia injury in hypertrophic myocardium. Cardiovasc Drugs Ther 2020; 34 (01) 3-14
- 10 Qi D, Atsina K, Qu L. et al. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest 2014; 124 (08) 3540-3550
- 11 Zhang C, Zhu HY, Wang J. et al. The mechanism of the inflammatory response of TCM in the prevention and treatment of cerebral ischemia-reperfusion injury based on the theory of blood circulation and blood stasis. J Zhejiang Chin Med Univ 2020; 44 (01) 107-110
- 12 Callens M, Kraskovskaya N, Derevtsova K. et al. The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer's disease. Biochim Biophys Acta Mol Cell Res 2021; 1868 (06) 118997
- 13 Lamb HM. Double agents of cell death: novel emerging functions of apoptotic regulators. FEBS J 2020; 287 (13) 2647-2663
- 14 Korshunova AY, Blagonravov ML, Neborak EV. et al. BCL2–regulated apoptotic process in myocardial ischemia–reperfusion injury (review). Int J Mol Med 2021; 47 (01) 23-36
- 15 Zhu X, Wang K, Jin Y. et al. Multiplexed fluorometric determination for three microRNAs in acute myocardial infarction by using duplex-specific nuclease and MoS2 nanosheets. Mikrochim Acta 2019; 187 (01) 15
- 16 Zhu WS, Guo W, Zhu JN. et al. Hsp90aa1: a novel target gene of miR-1 in cardiac ischemia/reperfusion injury. Sci Rep 2016; 6: 24498
- 17 Hao YL, Fang HC, Zhao HL. et al. The role of microRNA-1 targeting of MAPK3 in myocardial ischemia-reperfusion injury in rats undergoing sevoflurane preconditioning via the PI3K/Akt pathway. Am J Physiol Cell Physiol 2018; 315 (03) C380-C388
- 18 Gan J, Tang FMK, Su X. et al. microRNA-1 inhibits cardiomyocyte proliferation in mouse neonatal hearts by repressing CCND1 expression. Ann Transl Med 2019; 7 (18) 455
- 19 Hong T, Wei Y, Xue X. et al. A novel anti-coagulative nano complex in delivering miRNA-1 inhibitor against microvascular obstruction of myocardial infarction. Adv Healthc Mater 2020; 9 (11) e1901783
- 20 Tao L, Jia L, Li Y, Song C, Chen Z. Recent advances of adapter proteins in the regulation of heart diseases. Heart Fail Rev 2017; 22 (01) 99-107
- 21 Heinen A, Nederlof R, Panjwani P. et al. IGF1 treatment improves cardiac remodeling after infarction by targeting myeloid cells. Mol Ther 2019; 27 (01) 46-58
- 22 Qiao Y, Zhao Y, Liu Y. et al. miR-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun 2016; 477 (04) 541-547
- 23 Wang C, Liu W, Zhang X, Wang Y, Liu H, Li H. MEK/ERK signaling is involved in the role of VEGF and IGF1 in cardiomyocyte differentiation of mouse adipose tissue-derived stromal cells. Int J Cardiol 2017; 228: 427-434
- 24 Wang P, Gao R, Wu T. et al. Accumulation of endogenous adenosine improves cardiomyocyte metabolism via epigenetic reprogramming in an ischemia-reperfusion model. Redox Biol 2023; 67: 102884
- 25 Wang L, Niu X, Hu J. et al. After myocardial ischemia-reperfusion, miR-29a, and Let7 could affect apoptosis through regulating IGF-1. BioMed Res Int 2015; 2015: 245412
- 26 Wang R, Bao H, Zhang S, Li R, Chen L, Zhu Y. miR-186–5p promotes apoptosis by targeting IGF-1 in SH-SY5Y OGD/R model. Int J Biol Sci 2018; 14 (13) 1791-1799
- 27 Liu K, Ying Z, Qi X, Shi Y, Tang Q. MicroRNA-1 regulates the proliferation of vascular smooth muscle cells by targeting insulin-like growth factor 1. Int J Mol Med 2015; 36 (03) 817-824