
RSS-Feed abonnieren
DOI: 10.1055/s-0041-1735144
Efficient Synthesis and Docking Analysis of Selective CDK9 Inhibitor NVP-2


Abstract
Graphical Abstract
NVP-2 (1), a potent and selective inhibitor of cyclin-dependent kinase 9 (CDK9), showed potent antitumor activity in preclinical studies. In this work, we designed and adopted a convergent synthetic route to efficiently synthesize NVP-2 (1). The key intermediate (7) was synthesized from malononitrile (2) and 1-bromo-2-(2-bromoethoxy)ethane (3) by successive cyclization, reduction, nucleophilic substitution with 2-bromo-6-fluoropyridine, and Suzuki–Miyaura reaction with (5-chloro-2-fluoropyridin-4-yl)boronic acid. Another key intermediate (11) was synthesized from (S)-1-methoxypropan-2-ol (8) by reaction with TsCl, electrophilic substitution reaction with tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate, and then by deprotection of Boc. Finally, a substitution reaction by the key intermediates (7) and (11) to afford the target product NVP-2 (1). The reaction conditions of the whole synthesis process were simple and mild, free of harsh conditions such as the microwave reaction and dangerous reagents in the original patent, and realized the efficient synthesis of NVP-2. In addition, we analyzed the binding mode of NVP-2 in the active pocket of CDK9 to provide reasonable design ideas for subsequent discovery of novel CDK9 inhibitors.
#
Introduction
Cyclin-dependent kinase 9 (CDK9) mainly regulates gene transcription elongation and messenger RNA (mRNA) maturation, and has emerged as an attractive therapeutic target for many malignant tumors, including hematological malignancies and solid tumors.[1] [2] [3] [4] [5] Many CDK9 inhibitors have been developed or are currently being in various stages of clinical development for cancer treatment.[6] [7] [8] [9] [10] NVP-2 (1), chemical name “4-(((5′-chloro-2'-(((1R,4r)-4-(((R)-1-methoxypropan-2-yl)amino)cyclohexyl)amino)-[2,4'-bipyridin]-6-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile,” is a novel selective ATP-competitive CDK9 inhibitor developed by Novartis in 2011. NVP-2 displays a very potent CDK9/CycT inhibitory activity with an IC50 value of 0.51 nmol/L, and more than 1,000-fold selectivity over CDK1/CycB, CDK2/CycA, and CDK16/CycY.[11] In 2017, Winter et al found that NVP-2 significantly inhibited the proliferation of leukemia cells such as Jurkat, SKW-3, and SUP-T11, and induced the apoptosis of MOLT4 leukemia cells, suggesting the antitumor effect of NVP-2 in vitro.[11] NVP-2 may become a promising antitumor agent to enter clinical research.
To date, a few synthesis routes of NVP-2 and its key intermediates have been reported. However, there are some shortcomings, such as harsh reaction conditions, low yield, many side reactions, and potential safety hazards, in these reports that are not conducive to industrial production.[12] [13] Therefore, in this work, we first optimized the synthetic route to realize the efficient synthesis of NVP-2. Since the binding mode between NVP-2 and CDK9 protein has not yet been reported in the literature, we analyzed the docking mode of NVP-2 in the active pocket of CDK9 to provide reasonable design ideas for subsequent discovery of novel CDK9 inhibitors.
#
Results
Optimized Synthetic Route
According to the literatures, the synthesis strategy of NVP-2 is divided into two parts: the first is to synthesize the key intermediates 4-(((5′-chloro-2'-fluoro-[2,4'-bipyridyl]-6-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile (7) and (1r,4R)-N 1-((R)-1-methoxy-2-propyl)cyclohexane-1,4-diamine (11), and then intermediates 7 and 11 undergo substitution reaction to obtain the desired product NVP-2 (1). However, there are some shortcomings, such as harsh reaction conditions, low yields, side reactions, and potential safety hazards, in the reported synthetic route ([Fig. 1]).
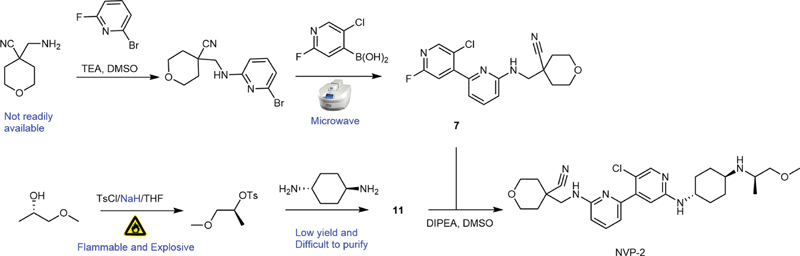
The synthetic route of key intermediate 7 has been optimized and improved on the basis of comprehensive relevant literature methods[12] [13]: tetrahydro-4H-pyran-4,4-dicarbonitrile (4) was synthesized from inexpensive and commercially available malononitrile (2) and 2,2'-dibromodiethyl ether (3) as starting materials under milder conditions. Intermediate 4 was readily reduced to 4-(aminomethyl)tetrahydro-2H-pyran-4-carbonitrile (5) using the mild reducing agent NaBH4. Nucleophilic substitution of 2-bromo-6-fluoropyridine by intermediate 5 occurs at high temperatures to give 4-(((6-bromopyridin-2-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile (6) with a high yield. Intermediate 6 and (5-chloro-2-fluoropyridin-4-yl)boronic acid under Suzuki–Miyaura reaction conditions gave the key intermediate 4-(((5′-chloro-2'-fluoro-[2,4'-bipyridyl]-6-yl)amino)methyl) tetrahydro-2H-pyran-4-carbonitrile (7). The synthetic route is shown in [Scheme 1]. In the improved synthetic route, we decreased the reaction temperature and appropriately extended the reaction time to efficiently obtain intermediate 4, thus overcoming the side reactions caused by the excessively high temperature. In the following step to synthesize intermediate 5, we could evade the forming of byproducts and improve the yield by slightly heating the reaction. Furthermore, in the Suzuki–Miyaura reaction, the harsh reaction conditions (high temperatures and microwave reactor) were used in the patent, which are not suitable for industrial manufacturing. We were able to avoid these harsh reaction conditions by changing the reaction solvent and employing an oil bath to effectively synthesize the key intermediate 7.

On the basis of the literature route,[12] [13] the synthetic route of key intermediate 11 is optimized and improved, shown in [Scheme 2]: inexpensive, readily available (S)-1-methoxy propan-2-ol (8) was used as the starting material, and reacted with p-toluenesulfonyl chloride to efficiently obtain (S)-1-methoxy-2-propyl-p-toluenesulfonate (9). The mixture of intermediate (9) and tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate was heated at reflux to efficiently obtain the intermediate tert-butyl ((1R,4r)-4-(((R)-1-methoxypropan-2-yl)amino)cyclohexyl)carbamate (10). Intermediate 10 was quickly reacted with hydrochloric acid solution to deprotect the Boc group, and gave (1r,4R)-N 1-((R)-1-methoxypropan-2-yl)cyclohexane-1,4-diamine (11) hydrochloride salt. Intermediate 11 and the above key intermediate 7 undergo substitution reaction at a higher temperature to obtain the final product NVP-2. In the synthetic route in the literatures, trans-cyclohexanediamine was directly reacted with intermediate 9 at high temperature to obtain intermediate 11.[12] [13] However, there are some disadvantages, such as byproduct formation, difficulties at purification, and low yield in this step. Also, the hazardous chemical reagent NaH was utilized in the synthesis of intermediate 9 in the literatures, posing significant safety concerns in industrial manufacturing. In our improved synthetic route, the raw material tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate was directly reacted with intermediate 9 at a mild temperature to efficiently obtain intermediate 10 (65% yield), which quickly deprotected the Boc group in the acid solution to obtain key intermediate 11 (95% yield). By our improved synthetic route, we can efficiently obtain the pure key intermediate 11. In addition, we used pyridine as the base instead of the dangerous reagent NaH, and greatly shortened the reaction time to efficiently synthesize the intermediate 9 with a high yield of 90%. In the last step to synthesize the NVP-2, the addition of inorganic base potassium carbonate could reduce the reaction time and increase the yield. During the entire NVP-2 reaction process, the whole operation is simple, efficient and safe, and apt to industrial production.

#
Docking Analysis
NVP-2 competitively binds to the ATP active pocket of CDK9 protein to exert potent CDK9 inhibitory activity. However, the binding mode between NVP-2 and CDK9 protein has not been reported. Revealing and predicting the binding mode between NVP-2 and CDK9 protein could provide reasonable design ideas for the discovery of novel CDK9 inhibitors. Therefore, we applied a computer docking technology to simulate the interaction between NVP-2 and CDK9 protein. The predicted binding mode of NVP-2 in the ATP active pocket of CDK9 protein is shown in [Fig. 2]. In the ATP active pocket of CDK9 protein ([Fig. 2A, B]), it contains a very important hinge region and some hydrophobic pockets. In the hinge region, Cys106 residue can form vital hydrogen bonds with many small-molecule CDK9 inhibitors, including flavopiridol, variolins, and others. The weakening or disappearance of this key hydrogen bond force will greatly reduce the inhibitory activity of CDK9 inhibitors. Asp104 is also an important amino residue located in the hinge region; some CDK9 inhibitors could form dual hydrogen bonds with Cys106 and Asp104 residues.[6] From the binding mode of NVP-2 in the CDK9 active pocket ([Fig. 2C, D]), it can be seen that the structure of NVP-2 can be well accommodated in the CDK9 active pocket, and the pyridine amino group in the NVP-2 structure can form dual hydrogen bonds with the Cys106 residue in the hinge region. This strong dual hydrogen bonding force endows NVP-2 with potent CDK9 inhibitory activity. In addition, the cyan group in the NVP-2 structure extends into the hydrophobic pocket of the CDK9 protein to enhance the interaction, while the cyclohexylamino fragment in the NVP-2 structure extends into the solvent-exposed region, suggesting that structural modification of this part can maintain or improve the inhibitory activity against CDK9. The above docking analysis provided valuable clues for modification and discovery of novel CDK9 inhibitors based on the structure of NVP-2.

#
#
Conclusion
NVP-2, a very potent and selective ATP-competitive CDK9 inhibitor, may be a promising antitumor agent to enter clinical research in the near future. In this work, we optimized the reagents and conditions of the synthetic route and realized the efficient synthesis of NVP-2. The whole synthesis process was simple and mild, and overcame the harsh conditions such as the microwave reaction and dangerous reagents. In addition, we analyzed the docking mode of NVP-2 in the active pocket of CDK9, and provided valuable clues for modification and discovery of novel CDK9 inhibitors based on the structure of NVP-2.
#
Experimental Section
Chemistry
Unless otherwise noted, all solvents and reagents were commercially available (Bidepharm, J&K Scientific, Sinopharm) and used without further purification. All reactions were monitored by thin-layer chromatography on 0.25 mm silica gel plates (60 GF-254) and visualized with UV light, or iodine vapor. 1H NMR and 13C NMR were generated in DMSO-d 6 or CDCl3 on Varian Mercury 400, 500, or 600 MHz NMR spectrometers. Chemical shifts were reported in parts per million (ppm). Multiplicity of 1H NMR signals was reported as single (s), double (d), triplet (t), quartet (q), and multiplet (m). ESI-MS (electrospray ionization mass spectra) were determined on an API 4000 spectrometer.
Synthesis of Dihydro-2H-pyran-4,4(3H)-dicarbonitrile (4)
A mixture of malononitrile (2, 7.0 g, 106 mmol), 1-bromo-2-(2-bromoethoxy)ethane (3, 24.57 g, 106 mmol)) in DMF (100 mL) was stirred in an ice bath for 20 minutes. Then to the reaction was added DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) (32.26 g, 212 mmol) dropwise. The reaction temperature was raised to 70°C and stirred for 3 hours. Then, the reaction was cooled to room temperature (r.t.), diluted with ethyl acetate, extracted with water and brine (150 mL × 3) until DMF was removed, and dried over anhydrous sodium sulfate. The organic phase was filtered and concentrated under reduced pressure to give a light brown solid (4, 12 g); yield: 83.18%. 1H NMR (600 MHz, DMSO-d 6) δ 3.73–3.67 (m, 4H), 2.32–2.26 (m, 4H).
#
Synthesis of 4-(Aminomethyl)tetrahydro-2H-pyran-4-carbonitrile (5)
To the solution of intermediate (4, 1.0 g, 7.34 mmol) in ethanol (35 mL) was slowly added sodium borohydride (0.83 g, 22.0 mmol) under ice bath conditions. The reaction mixture was removed from the ice bath and stirred at 60°C for 4 hours. The mixture was concentrated under reduced pressure and the resulting residue was diluted with ethyl acetate (100 mL), then washed with water and brine (150 mL × 3). The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated to dryness under a vacuum evaporator to give a crude product (5), which was used directly in the next step. Low-resolution mass spectrometry (LRMS) m/z calcd. for C7H12N2O [M + H]+ 141.09, found: 141.
#
Synthesis of 4-(((6-Bromo-pyridin-2-yl)amino)methyl)tetrahydro-2H-pyran-4-carboxylic carbonitrile (6)
To a solution of intermediate (5, 1.35 g, 9.61 mmol) in DMSO (10 mL) was consistently added 2-bromo-6-fluoropyridine (1.41 g, 8.01 mmol) and triethylamine (2.43 g, 20.9 mmol). The temperature of the oil bath was raised to 110°C, and the reaction was refluxed for 18 hours. Upon completion of the reaction, the reaction mixture cooled to ambient temperature, diluted with ethyl acetate, and washed with water and brine (150 mL × 3). After DMSO was washed and removed from the system, the organic layer was dried with anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography to give a light-yellow solid (6, 1.58 g); yield: 66.6%. LRMS m/z calcd. for C12H14BrN3O [M + H]+ 296.03, found: 296.2. 1H NMR (400 MHz, CDCl3) δ 7.24 (t, J = 7.8 Hz, 1H), 6.78 (d, J = 7.5 Hz, 1H), 6.39 (d, J = 8.2 Hz, 1H), 4.84 (t, J = 6.8 Hz, 1H), 4.00 (ddd, J = 12.4, 4.4, 2.2 Hz, 2H), 3.72–3.65 (m, 4H), 1.88 (dq, J = 13.8, 2.4 Hz, 2H), 1.77 (ddd, J = 13.8, 11.7, 4.4 Hz, 2H).
#
Synthesis of 4-(((5′-Chloro-2'-fluoro-[2,4'-bipyridyl]-6-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile (7)
A mixture of 6 (1.0 g, 3.38 mmol), 5-chloro-2-fluoropyridin-4-yl-boronic acid (0.888 g, 5.06 mmol), Pd(dppf)Cl2 (0.247 g, 0.38 mmol), Cs2CO3 (3.3 g, 10.13 mmol), and THF/H2O (2:1 [v/v], 20 mL) was stirred at 60°C for 5 hours. The reaction mixture was cooled to ambient temperature and concentrated in vacuo. The crude product was dissolved in ethyl acetate, washed with water and brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified by flash chromatography on silica gel to provide a light-yellow solid (7, 1.02 g); yield: 87.1%. LRMS m/z calcd. for C17H16ClFN4O [M + H]+ 347.10, found: 346.9. 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.21 (d, J = 2.4 Hz, 1H), 7.07 (d, J = 7.3 Hz, 1H), 6.57 (d, J = 8.3 Hz, 1H), 4.93 (s, 1H), 3.99 (ddd, J = 12.3, 4.5, 1.9 Hz, 2H), 3.78 (d, J = 6.8 Hz, 2H), 3.70 (td, J = 12.1, 1.9 Hz, 2H), 1.92 (dd, J = 13.7, 2.1 Hz, 2H), 1.76 (ddd, J = 13.8, 12.0, 4.5 Hz, 2H).
#
Synthesis of (S)-1-Methoxypropan-2-yl-4-methylbenzenesulfonate (9)
p-Toluene sulfonyl chloride (9.52 g, 49.9 mmol) in pyridine (20 mL) was stirred in an ice bath for 20 minutes, and to the reaction mixture was sequentially added (S)-1-methoxypropan-2-ol (8, 3.0 g, 33.3 mmol). The reaction vessel was sealed and stirred for 3 hours at 0°C. After 3 hours of stirring, the reaction mixture was diluted with water, then extracted with dichloromethane (150 mL × 3). The organic layer was combined and washed with 2 mol/L HCl (100 mL × 3), dried over anhydrous sodium sulfate, and concentrated in vacuo. The crude product was purified by column chromatography with a petroleum ether/ethyl acetate system to obtain a colorless oil (9, 8.1 g); yield: 99.6%.[1] H NMR (400 MHz, CDCl3) δ 7.87–7.69 (m, 2H), 7.55–7.37 (m, 2H), 4.66 (pd, J = 6.4, 3.7 Hz, 1H), 3.38–3.27 (m, 2H), 3.13 (s, 3H), 2.42 (s, 3H), 1.14 (d, J = 6.5 Hz, 3H).
#
Synthesis of tert-butyl((1S,4r)-4-(((S)-1-methoxypropan-2-yl)amino)cyclohexyl)carbamate (10)
To a mixture of intermediate 9 (1.0 g, 4.09 mmol) and K2CO3 (1.13 g, 8.19 mmol) in DMSO (20 mL) was slowly added tert-butyl ((1r,4r)-4-aminocyclohexyl)carbamate (1.32 g, 6.14 mmol). The remaining mixture was heated for 2 hours at 110°C. When the intermediate (9) was nearly completed indicated by thin layer chromatography (TLC) monitoring, the reaction was cooled to r.t., diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine (100 mL × 3), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified by flash chromatography silica gel column to give a pale-yellow solid (10, 0.76 g); yield: 65.0%. LRMS m/z calcd. for C15H30N2O3 [M + H]+ 287.23, found: 287.1.[1] H NMR (400 MHz, CDCl3) δ 4.38 (s, 1H), 3.41 (s, 1H), 3.34 (s, 3H), 3.29 (dd, J = 9.2, 4.5 Hz, 1H), 3.23 (dd, J = 9.3, 7.0 Hz, 1H), 3.01 (td, J = 6.7, 4.7 Hz, 1H), 2.51 (tt, J = 11.2, 3.9 Hz, 1H), 2.07–1.97 (m, 3H), 1.92 (dp, J = 9.3, 3.3 Hz, 1H), 1.44 (s, 9H), 1.24–1.09 (m, 4H), 1.03 (d, J = 6.4 Hz, 3H).
#
Synthesis of (1r,4R)-N 1-((R)-1-Methoxypropan-2-yl)cyclohexane-1,4-diamine (11)
To the solvent of intermediate 10 (1.0 g, 3.49 mmol) in DCM (5 mL) was slowly added 4 mol/L hydrochloric acid in dioxane (10 mL). Then the reaction was stirred at r.t. for 4 hours. The reaction mixture was concentrated in vacuo to give a white solid (11, 0.62 g); yield: 95.0%. LRMS m/z calcd. for C10H22N2O [M + H]+ 187.30, found: 187.1.1H NMR (400 MHz, CDCl3) δ 3.34 (s, 3H), 3.28 (dd, J = 9.2, 4.5 Hz, 1H), 3.22 (dd, J = 9.2, 6.9 Hz, 1H), 3.00 (td, J = 6.6, 4.7 Hz, 1H), 2.66 (ddt, J = 14.6, 9.5, 3.8 Hz, 1H), 2.49 (tt, J = 10.9, 3.9 Hz, 1H), 1.97 (dt, J = 12.3, 3.2 Hz, 1H), 1.93–1.83 (m, 3H), 1.26 (s, 2H), 1.17–1.12 (m, 2H), 1.02 (d, J = 6.4 Hz, 3H).
#
Synthesis of NVP-2 (1)
A mixture of intermediate 7 (0.5 g, 1.44 mmol), intermediate 11 (0.223 g, 1.73 mmol), and K2CO3 (0.598 g, 4.33 mmol) in DMSO (20 mL) was stirred at 110°C for 16 hours. When TLC monitoring showed the starting material was nearly completely consumed in the reaction, then the reaction was cooled to r.t., diluted with ethyl acetate, and washed with water (50 mL × 3). The organic phase was dried with anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude material was purified by flash column chromatography to obtain NVP-2 (1, 0.21 g) as a gray solid; yield: 28.4%. LRMS m/z calcd. for C27H37ClN6O2 [M + H]+ 513.27, found: 512.8. 1H NMR (500 MHz, DMSO-d6 ) δ 8.02 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.08 (t, J = 6.5 Hz, 1H), 6.78 (d, J = 7.2 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 6.61 (d, J = 11.7 Hz, 2H), 3.90 (ddd, J = 12.1, 4.5, 2.1 Hz, 2H), 3.67 (d, J = 6.4 Hz, 2H), 3.62–3.53 (m, 1H), 3.48 (td, J = 12.1, 2.0 Hz, 2H), 3.25 (s, 3H), 3.21–3.18 (m, 1H), 3.14 (dd, J = 9.2, 5.5 Hz, 1H), 2.92 (p, J = 6.1 Hz, 1H), 2.50–2.45 (m, 1H), 1.97–1.83 (m, 5H), 1.68 (ddd, J = 13.7, 11.8, 4.5 Hz, 2H), 1.26–1.02 (m, 5H), 0.94 (d, J = 6.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6 ) δ 158.26, 157.24, 152.31, 147.07, 146.75, 137.06, 122.45, 114.97, 112.40, 109.25, 108.58, 77.12, 63.85, 58.12, 52.76, 49.38, 48.47, 47.19, 38.62, 32.71, 32.41, 31.88, 31.32, 31.16, 18.15.
#
#
Docking Simulation
The binding mode between NVP-2 and the CDK9 active pocket was completed by the conventional computer docking software Schrödinger-Maestro (https://www.schrodinger.com/products/maestro, version 11.0, Schrödinger software package, New York, United States, 2020). The structural optimization of CDK9 protein (PDB number: 4BCF) was completed by the Protein Preparation Wizard sector in Maestro. The structure characterization of CDK9 protein was performed by adding hydrogens, creating disulfide bonds, filling in missing side chains, deleting water, restrained minimization, and other optimization operations (other parameters are kept unchanged). Subsequently, the receptor grid box (10 × 10 × 10 Å) was obtained based on the optimized structure of CDK9 protein. Continue to use LigPrep in Maestro to process the NVP-2 structure to generate various three-dimensional molecular configurations, and then used the Ligand Docking sector to complete the docking simulation between NVP-2 and CDK9 protein.
#
#
#
Conflict of Interest
All authors declare no conflict of interest.
# These authors contributed equally to this work.
-
References
- 1 Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24 (11) 1770-1783
- 2 Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov 2020; 10 (03) 351-370
- 3 Franco LC, Morales F, Boffo S, Giordano A. CDK9: a key player in cancer and other diseases. J Cell Biochem 2018; 119 (02) 1273-1284
- 4 Zhang H, Pandey S, Travers M. et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018; 175 (05) 1244.e26-1258.e26
- 5 Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (02) 130-146
- 6 Wu T, Qin Z, Tian Y. et al. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J Med Chem 2020; 63 (22) 13228-13257
- 7 Cho BC, Dy GK, Govindan R. et al. Phase Ib/II study of the pan-cyclin-dependent kinase inhibitor roniciclib in combination with chemotherapy in patients with extensive-disease small-cell lung cancer. Lung Cancer 2018; 123: 14-21
- 8 Mita MM, Joy AA, Mita A. et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 2014; 14 (03) 169-176
- 9 Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest New Drugs 2008; 26 (01) 59-65
- 10 Cidado J, Shen M, Grondine M. et al. Abstract 3572: AZ5576, a novel potent and selective CDK9 inhibitor, induces rapid cell death and achieves efficacy in multiple preclinical hematological models. Cancer Res 2016; 76 (14) 3572
- 11 Winter GE, Mayer A, Buckley DL. et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell 2017; 67 (01) 5.e19-18.e19
- 12 Olson CM, Jiang B, Erb MA. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol 2018; 14 (02) 163-170
- 13 Barsanti PA, Hu C, Jin J. et al. Heteroaryl compounds and their uses. U.S. Patent 8415381B2. April, 2013
Address for correspondence
Publikationsverlauf
Eingereicht: 08. Juni 2021
Angenommen: 07. Juli 2021
Artikel online veröffentlicht:
02. September 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution License, permitting unrestricted use, distribution, and reproduction so long as the original work is properly cited. (https://creativecommons.org/licenses/by/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24 (11) 1770-1783
- 2 Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov 2020; 10 (03) 351-370
- 3 Franco LC, Morales F, Boffo S, Giordano A. CDK9: a key player in cancer and other diseases. J Cell Biochem 2018; 119 (02) 1273-1284
- 4 Zhang H, Pandey S, Travers M. et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 2018; 175 (05) 1244.e26-1258.e26
- 5 Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (02) 130-146
- 6 Wu T, Qin Z, Tian Y. et al. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J Med Chem 2020; 63 (22) 13228-13257
- 7 Cho BC, Dy GK, Govindan R. et al. Phase Ib/II study of the pan-cyclin-dependent kinase inhibitor roniciclib in combination with chemotherapy in patients with extensive-disease small-cell lung cancer. Lung Cancer 2018; 123: 14-21
- 8 Mita MM, Joy AA, Mita A. et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 2014; 14 (03) 169-176
- 9 Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest New Drugs 2008; 26 (01) 59-65
- 10 Cidado J, Shen M, Grondine M. et al. Abstract 3572: AZ5576, a novel potent and selective CDK9 inhibitor, induces rapid cell death and achieves efficacy in multiple preclinical hematological models. Cancer Res 2016; 76 (14) 3572
- 11 Winter GE, Mayer A, Buckley DL. et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell 2017; 67 (01) 5.e19-18.e19
- 12 Olson CM, Jiang B, Erb MA. et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol 2018; 14 (02) 163-170
- 13 Barsanti PA, Hu C, Jin J. et al. Heteroaryl compounds and their uses. U.S. Patent 8415381B2. April, 2013