

Subscribe to RSS
DOI: 10.1055/s-0040-1714272
Immune-Mediated Coagulopathy in COVID-19 Infection
Authors
Viral infections, such as Epstein–Barr virus, Coxsackieviruses, and cytomegalovirus, are frequently blamed as a trigger for immune-mediated inflammation and autoimmunity, including the development of antiphospholipid syndrome. A variety of underlying mechanisms have been suggested, including virus direct overstimulatory effect on both innate and adaptive immune responses, cross-reactivity with self-antigens, and, lastly, direct effect on regulatory functions.[1] However, the high infectivity and damaging effect of coronavirus disease 2019 (COVID-19) on the respiratory tract, endothelial cells, and both innate and adaptive immunity are far beyond those of the vast majority of other viruses. While most patients with COVID-19 infection experience only mild fever, cough, myalgia, and weakness, within a few days some develop bilateral interstitial pneumonia and later acute respiratory distress syndrome, with many of them requiring long-lasting mechanical ventilation and intensive care.[2] The severity of respiratory failure and mortality in COVID-19-infected patients was reported to correlate with higher infectious dose followed by active and prolonged viral replication in pneumocytes, macrophages, and other immune cells. COVID-19 infection is characterized by a high incidence of thrombotic coagulopathies, up to the development of disseminated intravascular damage in some critical cases.[3] [4] However, the link between enhanced immune responses, endothelial damage, and coagulopathy in COVID-19-infected patients is yet to be clearly defined.
The Cytokine Storm
The cytokine storm is considered a unique event in coronavirus disease 2019 (COVID-19) infection and the main cause of ARDS and endothelial damage. Increased release of proinflammatory cytokines has been shown to occur in respiratory epithelial cells, dendritic cells, and macrophages at the early stage of coronavirus infection. At a later stage of disease, all of the aforementioned cells secrete low amounts of antiviral cytokines (interferons [IFNs]) along with high levels of proinflammatory cytokines such as interleukin (IL) 1, IL-6, tumor necrosis factor (TNF), and chemokines. The COVID-19-related cytokine storm is believed to be mainly a result of overwhelming innate immune responses. In this case, proinflammatory chemokines attract and activate neutrophils to secrete high levels of neutrophil extracellular traps, peroxidase and reactive oxygen species, and activate matrix metalloproteinases, which aggravate tissue damage in the lungs and cardiovascular system. In addition to the increased activation of T helper (Th) 1 cells and the production of relevant cytokines, COVID-19 has been reported to activate Th2 responses and increase the production of IL-4 and IL-10.[5] [6] Both cytokine types have been found to positively correlate with systemic morbidity and mortality. Accumulated macrophages may receive further stimulating signals through IFN receptors on their surface, leading to the production of more chemo-attractants and development of macrophage activation syndrome, one of the highly suggested mechanisms responsible for endotheliitis in patients with severe COVID-19 infection.[7]
Endothelial Cell Damage and Cardiovascular Involvement
Accumulating evidence points to the fact that COVID-19 infects endothelial cells (ECs) through angiotensin-converting enzyme 2 (ACE2), which is expressed on the EC surface.[8] The destructive effect of COVID-19 on ECs was mainly demonstrated in critically ill patients. Autopsies of COVID-19 patients displayed viral inclusion structures in ECs, and histological analyses revealed an accumulation of inflammatory cells in parallel with abundance of apoptotic bodies in the endothelium of most small vessels.[9] Increased EC apoptosis appeared to be related, at least in part, to IFN-signaling and overactivation of the Fas–Fas ligand pathway.
Immune-Mediated Coagulopathy in COVID-19
The cytokine storm in COVID-19 was reported to be a consequence of an imbalanced immune-mediated response. Progression of COVID-19 infection was associated with lymphopenia and a significant increase in neutrophils. The absolute numbers of CD4+ T cells and CD8+ T cells as well as B cells were all decreased in correlation with increased disease severity, whereas activation markers such as CD45RO+ and HLA-DR (human leukocyte antigen – DR isotype) on CD4+ and CD8+ T cells were augmented.[10] Lymphopenia was suggested to result from the redistribution of circulating lymphocytes and/or their depletion following activation. The amount of peripheral T regulatory cells was also decreased, potentially contributing to the expansion of activated CD4+ T cells and IFN-γ producing CD8+ T cells.[11] [12] As ACE2 is expressed weakly on lymphocytes,[13] membrane fusion and intracellular destruction are unlikely to be the main mechanism for lymphopenia. Severe cases of COVID-19 infection tend to have higher leukocyte counts and neutrophil/lymphocyte ratio in parallel with lymphopenia. Reduced numbers of lymphocytes and elevated levels of IL-6, TNF, plasmin, D-dimer, and ferritin are reported to occur in association with increased coagulopathy and mortality.[14] [15] At this stage, many aspects of immune-mediated disorders and cytokine storm in COVID-19 patients remain to be elucidated. We do not know whether autoimmunity is frequent in these patients, although some reports demonstrated the presence of antiphospholipids antibodies in these patients.[16] It is not clear if endothelial damage is related to the increased production of anti-endothelial antibodies or to the presence of immune complexes and complement activation, or to a combination of all these events.
Hypercoagulation in COVID-19 infection is demonstrated in case series, with laboratory markers of hypercoagulation reported to be associated both with venous and arterial thrombosis. In many COVID-19-infected patients, markers such as thrombin time, activated partial thromboplastin time (APTT), fibrin degradation, and high D-dimer and ferritin levels are found to be associated with increased incidence of thrombosis.[17] Whether COVID-19-related thrombosis somehow corresponds to the presence of antiphospholipid antibodies or other autoantibodies is still unknown. In one study, the development of thrombosis in three COVID-19 patients was found to be associated with the presence of anticardiolipin antibodies (aCL) and anti-β2-glycoprotein I antibodies (ab2GPI).[18] In another study, 50 COVID-19 patients were assessed for the presence of lupus anticoagulant (LAC) using dilute Russell viper venom time and sensitive APTT tests. Of the 50 patients, 25 were found to be LAC positive, with only 3 being aCL and ab2GPI positive.[19]
As mentioned previously, dysregulated immune responses, including MAS in COVID-19, mainly during late stages of disease, play a crucial role in the development of endothelial damage, microvascular permeability, and thrombosis. COVID-19 is also associated with moderate-to-severe thrombocytopenia, mainly reported in patients with other markers of immune-mediated inflammation.[20] The lower the platelet count, the higher the incidence of life-threatening disseminated intravascular coagulation (DIC) and mortality. This suggests that COVID-19-mediated enhanced immune responses may trigger platelet activation, leading to complex vascular changes through specific receptors and granule release.[20] [21]
Linking Cytokine Storm, Endotheliitis, and Coagulopathy
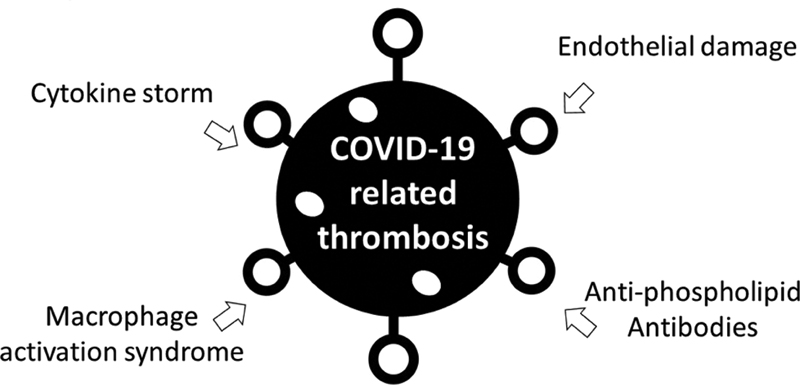
The pathogenesis of EC damage and DIC in small arterial branches of pulmonary arteries, cardiovascular, and mesenterium of COVID-19 patients is a complex multifactorial process. Related coagulopathy should be assessed and treated as immune-mediated rather than classical APS. The finding of positive antiphospholipid antibodies in some COVID-19-infected patients is insufficient to suggest that these antibodies are directly involved in COVID-19-related thrombosis. However, life-threatening microthrombosis and DIC in COVID-19 share similar clinical and laboratory features with catastrophic APS. In both, multiorgan involvement, including lungs, mesenteric arteries, and the cardiovascular system, is associated with severe morbidity. Likewise, proinflammatory cytokines, D-dimer, and ferritin are increased in association with coagulopathy ([Fig. 1]).
Conclusion
With the above in mind, COVID-19-infected patients should be given antiviral drugs as early as possible to maximally decrease the viral load. Anti-inflammatory drugs such as steroids, anti-IL-6, and hydroxychloroquine are often used to suppress the cytokine storm, and anticoagulation is employed in patients with more advanced disease. Many other therapeutic strategies are currently under evaluation, including intravenous immunoglobulins and (convalescent) plasma of cured patients enriched with specific anti-COVID-19 neutralizing IgG. However, multiple questions remain unanswered. What is the exact scenario of immune-mediated coagulopathy in COVID-19? How to predict, as early as possible, the course of COVID-19 worsening? Which are the best effective treatments based on higher level of evidence? Should routine hemostasis tests, such as D-dimer, prothrombin time, APTT, and fibrinogen, be used to guide the clinical decision-making? What value may other more specialized assays, including antiphospholipid antibody and LAC, have in COVID-19 evaluation?
Conflict of Interest
Dr. Brenner reports personal fees from Pfizer, LEO Pharma, Sanofi, ROVI Laboratories, and Bayer Pharmaceuticals, outside the submitted work.
-
References
- 1 Toubi E, Vadasz Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev 2019; 18 (03) 306-311
- 2 Henderson LA, Canna SW, Schulert GS. et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol 2020; (e-pub ahead of print)
- 3 Llitjos JF, Leclerc M, Chochois C. et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost 2020; (e-pub ahead of print)
- 4 Cao W, Li T. COVID-19: towards understanding of pathogenesis. Cell Res 2020; 30 (05) 367-369
- 5 Chen G, Wu D, Guo W. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 2020; 130 (05) 2620-2629
- 6 Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect 2020; 80 (06) 607-613
- 7 Li X, Xu S, Yu M. et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol 2020; (e-pub ahead of print)
- 8 Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020; 46 (04) 586-590
- 9 Varga Z, Flammer AJ, Steiger P. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020; 395 (10234): 1417-1418
- 10 Vardhana SA, Wolchok JD. The many faces of the anti-COVID immune response. J Exp Med 2020; 217 (06) e20200678
- 11 Wang F, Hou H, Luo Y. et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 2020; 5 (10) 137799
- 12 Zheng M, Gao Y, Wang G. et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 2020; 17 (05) 533-535
- 13 Xu H, Zhong L, Deng J. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 2020; 12 (01) 8
- 14 Qin C, Zhou L, Hu Z. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis 2020; (e-pub ahead of print)
- 15 Thachil J, Tang N, Gando S. et al. Laboratory haemostasis monitoring in COVID-19. J Thromb Haemost 2020; (e-pub ahead of print)
- 16 Hossri S, Shadi M, Hamarsha Z, Schneider R, El-Sayegh D. Clinically significant anticardiolipin antibodies associated with COVID-19. J Crit Care 2020; 59: 32-34
- 17 Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 2020; 18 (04) 844-847
- 18 Zhang Y, Xiao M, Zhang S. et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 2020; 382 (17) e38
- 19 Harzallah I, Debliquis A, Drénou B. Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost 2020; (e-pub ahead of print)
- 20 Terpos E, Ntanasis-Stathopoulos I, Elalamy I. et al. Hematological findings and complications of COVID-19. Am J Hematol 2020; 95 (07) 834-847
- 21 Liu Y, Sun W, Guo Y. et al. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets 2020; 31 (04) 490-496
Address for correspondence
Publication History
Article published online:
02 September 2020
© 2020. Thieme. All rights reserved.
Thieme Medical Publishers
333 Seventh Avenue, New York, NY 10001, USA.
-
References
- 1 Toubi E, Vadasz Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun Rev 2019; 18 (03) 306-311
- 2 Henderson LA, Canna SW, Schulert GS. et al. On the alert for cytokine storm: immunopathology in COVID-19. Arthritis Rheumatol 2020; (e-pub ahead of print)
- 3 Llitjos JF, Leclerc M, Chochois C. et al. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost 2020; (e-pub ahead of print)
- 4 Cao W, Li T. COVID-19: towards understanding of pathogenesis. Cell Res 2020; 30 (05) 367-369
- 5 Chen G, Wu D, Guo W. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 2020; 130 (05) 2620-2629
- 6 Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect 2020; 80 (06) 607-613
- 7 Li X, Xu S, Yu M. et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol 2020; (e-pub ahead of print)
- 8 Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020; 46 (04) 586-590
- 9 Varga Z, Flammer AJ, Steiger P. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020; 395 (10234): 1417-1418
- 10 Vardhana SA, Wolchok JD. The many faces of the anti-COVID immune response. J Exp Med 2020; 217 (06) e20200678
- 11 Wang F, Hou H, Luo Y. et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 2020; 5 (10) 137799
- 12 Zheng M, Gao Y, Wang G. et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 2020; 17 (05) 533-535
- 13 Xu H, Zhong L, Deng J. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 2020; 12 (01) 8
- 14 Qin C, Zhou L, Hu Z. et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis 2020; (e-pub ahead of print)
- 15 Thachil J, Tang N, Gando S. et al. Laboratory haemostasis monitoring in COVID-19. J Thromb Haemost 2020; (e-pub ahead of print)
- 16 Hossri S, Shadi M, Hamarsha Z, Schneider R, El-Sayegh D. Clinically significant anticardiolipin antibodies associated with COVID-19. J Crit Care 2020; 59: 32-34
- 17 Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 2020; 18 (04) 844-847
- 18 Zhang Y, Xiao M, Zhang S. et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 2020; 382 (17) e38
- 19 Harzallah I, Debliquis A, Drénou B. Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost 2020; (e-pub ahead of print)
- 20 Terpos E, Ntanasis-Stathopoulos I, Elalamy I. et al. Hematological findings and complications of COVID-19. Am J Hematol 2020; 95 (07) 834-847
- 21 Liu Y, Sun W, Guo Y. et al. Association between platelet parameters and mortality in coronavirus disease 2019: retrospective cohort study. Platelets 2020; 31 (04) 490-496