Keywords
heparin - heparin proteoglycan - antiplatelet - anticoagulant
Mast Cells As the Origin of Heparin
Mast Cells As the Origin of Heparin
Heparin, as used clinically, is one specific member of the glycosaminoglycan (GAG)
family, originates from intestinal mucosa and provides a protective anti-inflammatory,
antimicrobial, and antithrombotic surface against outer invaders. Heparin was originally
derived from liver as a phosphatide, which turned out to possess anticoagulant activity.[1] Heparin has served for more than 75 years as a source material for modern anticoagulation.
The birth of heparin in the United States by the research of Jay MacLean and William
Henry Howell has offered a unique and long-lasting clinical tool to manage and prevent
thrombosis.[1] Heparin is the preferred anticoagulant to prevent contact activation during certain
procedures, such as heart and vascular surgery and plasma exchange. The structural
studies for the first clinical use of heparin were performed in 1935 by a Finnish
doctor, Erik Jorpes, who had moved in 1919 from Finland to Stockholm Karolinska Institute.[2] An anecdote of his fanaticism for this material is written in his biography by Runar
Beckman: “When his wife was pregnant and the delivery was near some problems arose.
Jorpes was working late in the evenings in the laboratory, and when a friend asked
him to come home to help his wife he responded that babies are born all the time but
heparin is born only once.”[3]
The unfractionated heparin (UFH) is isolated from porcine or bovine intestinal mucosa
and is used in pharmaceutical manufacturing processes as active product ingredient.
It is further chemically or enzymatically degraded to lower molecular weight species,
which has been the mainstay of acute anticoagulation needs for several decades.
At the time of writing this article, Seminars in Thrombosis & Hemostasis (STH) contains, according to PubMed, as many as 592 contributions where heparin is mentioned.
The oldest entry dates back to 1975, by Gallus et al, “Thrombolysis with a Combination
of Small Doses of Streptokinase and Full Doses of Heparin”).[4] During the years, many insights into the heparin molecule, its structure and function,
as well as laboratory measures and clinical management have been provided by the journal.
The history of heparin and STH has shared parallel and complementary routes. The low-molecular-weight heparin, its
subcutaneous administration, and even the ultralow molecular species and the antithrombin-binding
pentasaccharide and entities without anticoagulant activities have been vividly discussed
during the past 40 years. Therefore, it is a great privilege for us to write this
article and propose the parent heparin proteoglycan as a model of future drug development.
The GAG surface is typical of all vasculature and organs. The glycocalyx delineates
endothelial and platelet surfaces and protects them from simple contact activation.
This natural barrier needs to be disrupted in order to initiate coagulation activity,
which according to its physiology localizes and limits the action to the site of injury.
Under severe coagulation disorders, such as disseminated intravascular coagulation
and thrombotic thrombocytopenic purpura, this protective function is lost or altered
and generalized thrombosis occurs. For this purpose, the endothelial glycocalyx and
the heparan sulfate provide the scaffold to support the potent anticoagulative roles
of several molecular interactions, such as antithrombin, tissue factor pathway inhibitor,
protein C–S system, heparin cofactor II, fibrinolysis, and protection against complement
system–mediated activation and fostering regulatory factor H function.
Mast cells are an integral component of the inflammatory triad, together with neutrophils
and lymphocytes. Mast cells reside in tissues, in the peritoneal cavity, in the lungs
and vasculature, where they in particular surround the outer surface of vessels, in
the adventitial layers. This is the very same site where the strongest tissue factor
activity is expressed. The deeper the damage, the more coagulation activity occurs
within a given vessel. Thus, the regulatory steps for this activation need to be adopted
accordingly. The tissue-associated mast cells upon their activation release heparin
and chondroitin proteoglycans (HEP-PG and CS-PG, respectively). HEP-PG are large,
on average, 750 kDa structures, containing over 10 heparin chains, of various lengths
(likely 20–75 kDa) bound to serine–glycine backbone. This heparin structure also supports
the activity of enzymes, such as chymase and tryptase, which both exert anticoagulant
properties, such as inhibition of thrombin.[5]
[6] Moreover, mast cells contain plasminogen activators, being simultaneously devoid
of their regulators of plasminogen-activator inhibitors (PAI-1 and PAI-2), as opposed
to all other known cell types.[7]
HEP-PG Are Strong Inhibitors of Platelet Aggregation and Deposition to Collagen
HEP-PG Are Strong Inhibitors of Platelet Aggregation and Deposition to Collagen
We isolated serosal mast cells and noted that when activated these release the HEP-PG,
which then exhibits a strong inhibitory action against platelet aggregation induced
by collagen.[8] UFH has been used as the control for HEP-PG in all these experiments. Also, under
high shear rate blood flow over collagen, the platelet deposition is significantly
reduced. HEP-PG attenuate platelet–collagen interactions under blood flow via von
Willebrand factor (VWF) and platelet glycoprotein (GP)IIb/IIIa-mediated mechanisms.
When chondroitinase-treated HEP-PG was used, the inhibitory capacity was maintained,
whereas heparinase eliminated the platelet inhibitory reactions of HEP-PG. HEP-PG
also arrests platelet thrombus growth at the immobilized collagen surface in vitro
and vascular injury sites in vivo.[9]
[10] The inhibition of platelet deposition and thrombus growth was evidenced both in
solution when spiked in whole blood and upon immobilization on the collagen surface.
Indeed, when immobilized directly to the collagen surface, platelet deposition was
diminished. In these experiments UFH was significantly more potent anticoagulant via
antithrombin than HEP-PG, whereas HEP-PG exerted its anticoagulant action via heparin
cofactor II (Coenraad Hemker, MD, PhD, and Suzette Beguin, MD, PhD, unpublished data,
1998). In the in vivo experiments, the locally administered HEP-PG at the site of
rat femoral artery anastomosis maintained patency while saline and UFH failed to do
so.[10]
The concept of mast cell-derived tissue regulation of coagulation was studied in human
bleeding time (BT) wounds, where allergen-induced challenge in the BT wound prolonged
the BT over the period of follow-up.[11] The prolongation of BT started at 60 minutes and was maximal at 120 minutes after
the allergen challenge in comparison with the histamine control. Also, lesions of
cutaneous mastocytoma patients when subjected to BT wound expressed prolonged BT from
the injury site.
In a rodent study of vascular anastomosis, the local administration of UFH versus
HEP-PG clearly showed that the HEP-PG could maintain the vessel patency, and the 111-Indium–labeled–infused
platelets deposited less to the anastomosis surface with the exposed collagen and
vascular matrix in the presence of HEP-PG than in the presence of UFH.[10] Rat femoral arteries were cross-sectioned and the anastomosis was created in a regular
manner. Before closing the last sutures the inner vessel surface was flushed with
a small volume of solution with HEP-PG and the blood flow was allowed to enter the
vessel. In [Fig. 1], the uncovered anastomosis surface can be recognized with the visible stitches,
whereas in the controls large thrombi were encountered on the vascular anastomosis
sites. The HEP-PG has also been shown to carry significant antiproliferative properties
in vascular smooth muscle cells, exceeding that of pharmaceutical heparin species.[12]
 Fig. 1 Scanning electron micrograph of an anastomosis site in a rat femoral artery when
either UFH (A) or HEP-PG (B) had been administered locally by flushing (at 100 µg/mL)
with the solution before sacrificing the animal. The anastomosis site was opened for
tissue preservation. HEP-PG, heparin proteoglycans; UFH, unfractionated heparin. (Reprinted
with permission from Olsson E, Asko-Seljavaara S, Lassila R. Topically administered
macromolecular heparin proteoglycans inhibit thrombus growth in microvascular anastomoses.
Thromb Haemost 2002;87:245–251.)
Fig. 1 Scanning electron micrograph of an anastomosis site in a rat femoral artery when
either UFH (A) or HEP-PG (B) had been administered locally by flushing (at 100 µg/mL)
with the solution before sacrificing the animal. The anastomosis site was opened for
tissue preservation. HEP-PG, heparin proteoglycans; UFH, unfractionated heparin. (Reprinted
with permission from Olsson E, Asko-Seljavaara S, Lassila R. Topically administered
macromolecular heparin proteoglycans inhibit thrombus growth in microvascular anastomoses.
Thromb Haemost 2002;87:245–251.)
HEP-PG Mimetics APACs As Dual Antiplatelet and Anticoagulants
HEP-PG Mimetics APACs As Dual Antiplatelet and Anticoagulants
We have constructed semisynthetic molecule complexes which can be tailored to contain
a desired amount of heparin chains and they express both anticoagulant and antiplatelet
actions, especially against platelet aggregation induced by collagen. The platelet
inhibition becomes a highly specific feature, which the soluble unfractionated and
low-molecular-weight heparin species do not possess at clinically relevant or even
higher concentrations. In contrast, heparin has in fact been shown also to exert some
direct activation on platelets as well as the well-known immunological complications
of heparin-induced thrombocytopenia and thrombosis.[13]
[14]
[15] APACs (antiplatelet and anticoagulant constructs) are conjugates with dual antiplatelet
and anticoagulant properties. We will report the antithrombotic effects of APACs in
vitro, and in rat vascular anastomosis and baboon thrombosis models in vivo.
Methods
Agonist-induced (collagen, Nycomed Pharma, Munich Germany; Chrono-Par, Chronolog
Corp., Havertown, PA; ristocetin, Helena Biosciences, Gateshaed, United Kingdom;
ADP, Sigma-Aldrich, St Louis, MO) platelet aggregation was assessed in citrate anticoagulated
blood in platelet-rich plasma (PRP) (Aggram, Helena Biosciences). The anticoagulant
effects of APAC and UFH were compared using activated partial thromboplastin time
(aPTT),[16] prothrombin time (PT), thrombin time, anti-FXa activity, fibrinogen, FVIII:C, VWF
activity and antigen, and calibrated automated thrombogram (CAT)[17]
[18] measurements. PT and aPTT were always measured using Thromborel-S and Actin FSL
reagents (both from Siemens Inc., Germany). In CAT, platelet-poor plasma (PPP) was
supplemented with 5 pM tissue factor (TF) and PRP with 1 pM TF (PPP reagent and PRP
reagent, respectively, Stago, France).
The intravenously administered anticoagulant efficacy and safety of APACs, UFH, and
vehicle (saline) were compared in a rat tail BT model.[19]
[20] In brief, BT experiments were performed under general anesthesia (2% isoflurane-O2)
and study compounds were administered and blood was collected via femoral vein catheter.
Before dosing, blood was collected in 3.2% sodium citrate for aPTT and PT analysis.
APACs, UFH or the vehicle were injected within 30 seconds. At 5 minutes a blood sample
was collected to measure aPTT and control the level of anticoagulation. At 10 minutes
the tail was cut at 5 mm from the distal tip with a scalpel and immersed in warm (35 ± 2°C)
0.9% saline. The BT was recorded until the bleeding had been ceased at least for 5
to 10 seconds.
The antithrombotic efficacy of APACs and UFH in comparison with vehicle was also studied
in two well established models of acute thrombosis in baboons. In a modified Folts
model an extracorporeal AV-shunt was created between the femoral artery and vein in
an anesthetized animal.[21]
[22] The blood flow was controlled by an external constrictor placed on the artery and
the flow was monitored with a probe. The artery was injured from outside by cross-clamping
twice for 10 seconds with a Martin needle holder (Hegar-Baumgartner TC Gold 14 cm).
All side branches in the proximity of the injury were ligated. The shunt was punctured
with a needle (26 G) 1 cm proximal to the vascular access for injecting a bolus (4
mg/mL) of APAC, UFH or saline. The injury was treated for 3 minutes with the study
substance before being exposed to blood flow. Immediately after recovering the baseline
blood flow, an external constrictor was placed on the injury and flow was reduced
to 30 to 100 mL/min (reflecting a stenosis of 90 to 30%). The accumulation of platelets
on the stenosed artery was detected by the reduced blood flow and recorded as cyclic
flow reductions (CFR). At 5 mL/min the artery was considered occluded and the thrombus
was dislodged by releasing the constrictor and flushing with saline. After baseline
blood flow was recovered, stenosis was reapplied and the experiment repeated.
In the second baboon model, thrombosis was induced by placing collagen-coated polytetrafluoroethylene
grafts (2 cm, 4-mm lumen) into an externalized arteriovenous shunt.[23] The thrombogenic collagen surface was treated for 10 minutes with APAC or UFH (both
at 4 mg/mL). Blood flow was initiated (100 mL/min; a shear rate of 265/s) and the
deposition of 111-Indium-labeled platelets and fibrin (accumulation of 125-Iodine-fibrinogen)
was quantified for 60 minutes in an anesthetized animal. Efficacy, distribution, and
retention on-site of locally administered 64-Cu-labeled APAC or UFH (3 mg/kg) were
assessed by positron emission tomography (PET) imaging for 50 hours of partially ligated
(two loose sutures 1 cm apart) femoral artery anastomoses in rats.
APAC Inhibits Collagen-Induced Platelet Aggregation with Simultaneous Anticoagulant
Efficacy In Vitro
APAC Inhibits Collagen-Induced Platelet Aggregation with Simultaneous Anticoagulant
Efficacy In Vitro
APAC inhibited platelet aggregation in human PRP induced by collagen and ristocetin
(the latter requiring about a 10-fold higher concentration of APAC) in contrast to
UFH, but not by ADP. The difference between the doses to reach at least 70% inhibition
of platelet aggregation, varied between 3 to 90 µg/mL in collagen-induced platelet
aggregation in citrate anticoagulated PRP between the blood donors ([Fig. 2]). The mean ED50 dose to inhibit collagen-induced aggregation was approximated to
30 µg/mL (n = 12; independent donors; inhibition 54 ± 24%, mean and SD).
 Fig. 2 Inhibition of collagen-induced maximal platelet aggregation in the presence of APAC
in citrate anticoagulated PRP with three different donors: A high (open circle), a
moderate (open square), and low (open diamond) responder to APAC. The mean inhibition
in all three donors in the presence of UFH (black triangle) is shown. Inhibition of
the maximal platelet aggregation relative to the vehicle (PBS) is shown as a percentage
(%). APAC, antiplatelet and anticoagulant; PBS, phosphate-buffered saline; PRP, platelet-rich
plasma; UFH, unfractionated heparin.
Fig. 2 Inhibition of collagen-induced maximal platelet aggregation in the presence of APAC
in citrate anticoagulated PRP with three different donors: A high (open circle), a
moderate (open square), and low (open diamond) responder to APAC. The mean inhibition
in all three donors in the presence of UFH (black triangle) is shown. Inhibition of
the maximal platelet aggregation relative to the vehicle (PBS) is shown as a percentage
(%). APAC, antiplatelet and anticoagulant; PBS, phosphate-buffered saline; PRP, platelet-rich
plasma; UFH, unfractionated heparin.
The anticoagulant effects of APAC measured by aPTT and thrombin time were comparable
or even somewhat better than UFH, while FVIII, fibrinogen, and VWF variables remained
unaltered (results not shown) at an equal heparin concentration in vitro. The anticoagulant
action of APAC was neutralized by protamine sulfate to a similar manner as with UFH
(data not shown).
In the presence of platelets, APAC molecules dose-dependently impaired thrombin generation
by initially prolonging the lag time and time to peak (ttpeak), while reducing the
peak and endogenous thrombin potential (ETP) both in citrate anticoagulated PPP (supplemented
with 5 pM TF and triggered with Ca2+) and PRP (supplemented with 1 pM TF and triggered with Ca2+). In the representative CAT analysis in PRP ([Fig. 3A–C]), the baseline (phosphate buffered saline; PBS) lag time was 9 minutes, ttpeak 20
minutes, peak 58 nM, and ETP 965 nM. At 0.5 µg/mL, UFH prolonged the lag time 45%,
ttpeak 48% and reduced the peak 11% in comparison with the baseline. At 0.5 µg/mL,
APAC prolonged the lag time by 70%, ttpeak by 134%, and reduced the peak by 30% in
comparison with the baseline ([Fig. 3A]). Overall, APAC prolonged the lag time 1.6-fold, ttpeak 2.8-fold, and reduced the
peak 2.7-fold more than UFH. At 1.0 µg/mL UFH prolonged the lag time 75%, ttpeak 159%,
and reduced the peak 41%, while in contrast, already at 1.0 µg/mL APAC abolished the
thrombin generation ([Fig. 3B]). At 1.5 µg/mL, UFH prolonged the lag time 79%, while the ttpeak and peak were immeasurable
([Fig. 3C]). ETP was not determined for UFH and APAC due to the significantly delayed thrombin
generation at heparin concentrations of 0.5 to 1.5 µg/mL.
 Fig. 3 Reduced thrombin generation in PRP in the presence of APAC compared to UFH using
CAT. The thrombograms in the presence of vehicle (PBS) and of (A) 0.5 µg/mL, (B) 1.0
µg/mL and (C) 1.5 µg/mL of APAC or UFH in citrate anticoagulated PRP supplemented
with 1 pM TF are shown. APAC, antiplatelet and anticoagulant; CAT, calibrated automated
thrombogram; PBS, phosphate-buffered saline; PRP, platelet-rich plasma; UFH, unfractionated
heparin.
Fig. 3 Reduced thrombin generation in PRP in the presence of APAC compared to UFH using
CAT. The thrombograms in the presence of vehicle (PBS) and of (A) 0.5 µg/mL, (B) 1.0
µg/mL and (C) 1.5 µg/mL of APAC or UFH in citrate anticoagulated PRP supplemented
with 1 pM TF are shown. APAC, antiplatelet and anticoagulant; CAT, calibrated automated
thrombogram; PBS, phosphate-buffered saline; PRP, platelet-rich plasma; UFH, unfractionated
heparin.
APAC in a Rat Tail BT Model and Acute Models of Thrombosis in Primates
APAC in a Rat Tail BT Model and Acute Models of Thrombosis in Primates
APAC variables APAC1 and APAC2 (with double coupling efficacy of heparin compared
to APAC1), and the UFH prolonged the aPTT and rat tail BT (n = 5–8) in comparison with the saline-treated animals. aPTT was prolonged at a dose
of 0.26 mg/kg by eightfold with APAC1 but only doubled with APAC2 over the baseline
([Table 1]). At 0.96 mg/kg aPTT prolonged by 10-fold with both APACs while UFH had already
reached the maximal time measured (180 seconds). At 1.2 mg/kg, also APAC1 induced
the maximal prolongation of aPTT while APAC2 clearly (12-fold) prolonged aPTT, which
still could be measured. PT was modestly elevated in all treatment groups (range ,
10–13 seconds) in comparison to the baseline level (9 seconds). Blood cell counts
were and remained normal.
Table 1
Comparison of APAC1 and APAC2 efficacy on hemostasis in a rat tail bleeding time (BT)
model
|
Treatment
mg/kg
|
BT
Mean (s) ± SEM
|
n
|
aPTT postdose
Mean (s) ± SEM
|
n
|
|
Saline
|
360.00 ± 54.79[a]
|
6
|
15.02 ± 0.26
|
6
|
|
UFH 0.96
|
1,949.00 ± 276.96[b]
|
6
|
180.00 ± 0.00
|
5
|
|
APAC1 0.26
|
619.17 ± 63.085[a]
[b]
|
6
|
119.14 ± 37.31
|
5
|
|
APAC1 0.96
|
986.29 ± 142.04[a]
[b]
|
7
|
150.20 ± 17.56
|
8
|
|
APAC1 1.2
|
2,467.50 ± 130.58[b]
|
6
|
180.00 ± 0.00
|
6
|
|
APAC2 0.26
|
803.17 ± 186.692[a]
[b]
|
6
|
27.50 ± 3.11
|
6
|
|
APAC2 0.96
|
790.67 ± 128.45[a]
[b]
|
6
|
149.10 ± 15.43
|
6
|
|
APAC2 1.2
|
1,704.17 ± 264.26[b]
|
6
|
174.83 ± 5.17
|
6
|
Abbreviations: APAC, antiplatelet and anticoagulant; SEM, standard error of mean;
UFH, unfractionated heparin.
Note: Saline was used as a negative control and unfractionated heparin (UFH) as the positive
control. Samples for activated partial thromboplastin time (aPTT) were collected at
5 minutes after dosing. Detection of bleeding time (BT) was initiated at tail cut
10 minutes after dosing. aPTT was measured up to 180 seconds. The average BT(s) and
the aPTT(s) with the standard error of the mean are shown. At 0.96 mg/kg UFH versus
antiplatelet and anticoagulant (APAC) 1 and APAC2, p values were 0.004 and 0.008, respectively. APACs showed shorter BTs than UFH despite
of the relatively equal aPTT prolongation.
a Statistically different from the UFH group, with p ≤ 0.05 following Student t-test.
b Statistically different from the vehicle group.
At 0.96 mg/kg UFH (approximately 1.5 IU/mL, a high clinically reachable level) prolonged
the tail BT over fivefold the baseline ([Table 1]). At 0.26, 0.96, and 1.2 mg/kg, APAC1 prolonged BT by 1.7-, 3-, and 7-folds the
baseline, respectively. APAC1 expressed shorter BT from the tail wound than UFH at
0.96 mg/kg. At both 0.26 and 0.96 mg/kg APAC2 prolonged the BT 2-fold and at 1.2 mg/kg
by 4.7-fold in comparison with the baseline. At both 0.96 and 1.2 mg/kg APAC2 exhibited
even 20 to 30% shortened BT than APAC1.
In the acute thrombosis models in baboons APAC and UFH were administered locally on
the injury at 4 mg/mL ([Fig. 4]). In the presence of UFH, the artery repeatedly (5 CFRs/27 min) occluded at a flow
rate of 100 mL/min (i.e., stenosed by 30%). In contrast, APAC effectively inhibited
thrombus formation until the experiment was interrupted, after the follow-up time
of 120 minutes. At the end of the experiment, at time point 180 minutes, the artery
was restenosed first to 50 mL/min (60% stenosis) and finally to 30 mL/min (90% stenosis),
while the local APAC continued to inhibit the occlusion for the selected test periods
(10 and 15 minutes, respectively). In the control experiments with UFH and saline
repetitive CFRs ensued (results not shown).
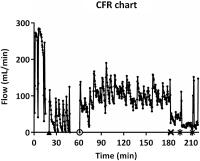 Fig. 4 A chart of CFRs after the local application of UFH and APAC1 (both at 4 mg/mL; 2 mg
in total) into the fresh injury site in the modified-Folt model of acute thrombosis
in baboons. Immediately after the baseline blood flow returned, the artery was stenosed
(30%) to the flow rate of 100 mL/min. Repeated occlusion (5 CFRs within 25 minutes)
was seen at the injury site treated with UFH (black triangle). In comparison with
APAC treatment (open circle) the fresh injury site remained open for the duration
of the experiment: First for 120 minutes at the arterial blood flow of 100 mL/min
(open circle), secondly for 14 minutes with tightened stenosis at the arterial blood
flow of 50 mL/min (black cross), and finally for 10 and 15 minutes sequential periods
at harsh stenosis at blood flow of 30 mL/min (black stars). APAC, antiplatelet and
anticoagulant; CFR, cyclic flow reduction; UFH, unfractionated heparin.
Fig. 4 A chart of CFRs after the local application of UFH and APAC1 (both at 4 mg/mL; 2 mg
in total) into the fresh injury site in the modified-Folt model of acute thrombosis
in baboons. Immediately after the baseline blood flow returned, the artery was stenosed
(30%) to the flow rate of 100 mL/min. Repeated occlusion (5 CFRs within 25 minutes)
was seen at the injury site treated with UFH (black triangle). In comparison with
APAC treatment (open circle) the fresh injury site remained open for the duration
of the experiment: First for 120 minutes at the arterial blood flow of 100 mL/min
(open circle), secondly for 14 minutes with tightened stenosis at the arterial blood
flow of 50 mL/min (black cross), and finally for 10 and 15 minutes sequential periods
at harsh stenosis at blood flow of 30 mL/min (black stars). APAC, antiplatelet and
anticoagulant; CFR, cyclic flow reduction; UFH, unfractionated heparin.
In the baboon thrombosis model on the extracorporeal collagen graft, APAC reduced
platelet deposition on collagen by 34 ± 13% (n = 4, p = 0.01) in comparison with UFH. The distal thrombus propagation was also diminished
by 63 ± 11% (n = 4, p = 0.19) ([Fig. 5]). Results with UFH were similar to untreated control values (n = 21). Fibrin accumulation was reduced by APAC (45 ± 14%, n = 4), but not by UFH (1.1 ± 0.1%, n = 4, p = 0.001).
 Fig. 5 Comparison of APAC1 and UFH (both at 4 mg/mL) in collagen-induced thrombus formation
in a baboon model (n = 4). Reduced platelet deposition was seen in (A) collagen surfaces at the site of
application where the platelet on deposition was reduced in the presence of APAC1
by 34 ± 13% (SD; ± 6% SEM) in comparison with UFH (p = 0.01) and (B) thrombus that propagated 10 cm distal to the collagen segment where
the platelet deposition was reduced at the presence of APAC1 by 63 ± 11% (SD; ± 5%
SEM) in comparison with UFH (p = 0.19). APAC, antiplatelet and anticoagulant; SD, standard deviation; SEM, standard
error of mean; UFH, unfractionated heparin.
Fig. 5 Comparison of APAC1 and UFH (both at 4 mg/mL) in collagen-induced thrombus formation
in a baboon model (n = 4). Reduced platelet deposition was seen in (A) collagen surfaces at the site of
application where the platelet on deposition was reduced in the presence of APAC1
by 34 ± 13% (SD; ± 6% SEM) in comparison with UFH (p = 0.01) and (B) thrombus that propagated 10 cm distal to the collagen segment where
the platelet deposition was reduced at the presence of APAC1 by 63 ± 11% (SD; ± 5%
SEM) in comparison with UFH (p = 0.19). APAC, antiplatelet and anticoagulant; SD, standard deviation; SEM, standard
error of mean; UFH, unfractionated heparin.
In the rat anastomosis model about 10% APAC attached directly to the vascular application
site ([Fig. 6]). Compatible with the strong retention potential and slow degradation, PET detected
nondegraded APAC at the anastomotic sites in rats for over 50 hours (up to 120 hours),
whereas UFH was undetectable already after 24 hours (n = 2). APAC and UFH were cleared via urinary pathway.
 Fig. 6 An example of PET/CT images of the rat at 30 minutes, 6, 21, and 48 hours time points
following the local administration of 64-Copper-APAC2 at the site of anastomosis.
Remaining radioactivity is shown at the As. PET acquisition was performed on a Triumph
PET/CT scanner (Gamma Medica-Ideals, Nortridge, CA). The radioactivity remaining at
the As and in the organs was quantified using three-dimensional ROI. Quantitative
values at the As are expressed as the percentage-radioactivity relative to the first
measurement and corrected for the physical decay of the radiolabel. APAC, antiplatelet
and anticoagulant; As, application site; CT, computed tomography; PET, positron emission
tomography; ROI, regions of interest.
Fig. 6 An example of PET/CT images of the rat at 30 minutes, 6, 21, and 48 hours time points
following the local administration of 64-Copper-APAC2 at the site of anastomosis.
Remaining radioactivity is shown at the As. PET acquisition was performed on a Triumph
PET/CT scanner (Gamma Medica-Ideals, Nortridge, CA). The radioactivity remaining at
the As and in the organs was quantified using three-dimensional ROI. Quantitative
values at the As are expressed as the percentage-radioactivity relative to the first
measurement and corrected for the physical decay of the radiolabel. APAC, antiplatelet
and anticoagulant; As, application site; CT, computed tomography; PET, positron emission
tomography; ROI, regions of interest.
Discussion
APAC molecules show unique dual and localized antiplatelet and anticoagulant action
uniformly in several in vitro and in vivo experiments. In addition to inhibition of
platelet aggregation by collagen and anticoagulation (prolongation of aPTT and thrombin
time) in human PRP and PPP, in the absence of any systemic anticoagulation, APACs
were efficient with local administration in vivo in two baboon models of acute arterial
thrombosis and collagen-induced platelet deposition and fibrin formation. APACs may
be beneficial under several vascular applications where localized thrombosis continues
to be a problem despite the use of current antithrombotic medications. These current
agents are systemically administered and also pose a bleeding hazard, which may also
be lessened by the local drug administration due to a low concentration but prolonged
action. We obviously need to demonstrate that there is no significant leakage to the
tissue and that the vascular healing continues in a physiological manner. Our preliminary
experiments after 2 weeks of local application support intact healing and vascular
structures at the site of anastomosis, which has been treated with our APAC compounds.
In vitro CAT experiments in the presence and absence of platelets in plasma continue
to indicate the dual anticoagulant effect and inhibition of platelet-dependent procoagulant
action of APACs. The anticoagulant effects of APACs were comparable with UFH at similar
dose levels in vitro as previously reported.[24] In the presence of platelets, APAC impaired thrombin generation initially by prolonging
the lag time and ttpeak clearly more than equal doses of UFH. In sharp contrast to
UFH, APAC inhibited the collagen-induced platelet aggregation in PRP, but similar
to UFH, APAC had no effect on ADP-induced platelet aggregation in PRP.
In vivo BT experiments in rats showed a systemic aPTT prolongation compatible with
UFH, but again in deviation from UFH shortened BT, despite the dual anticoagulant
and antiplatelet in vitro action. The BT method of rat tail transection, where the
tail is immersed in warm saline, is uniformly used to study the anticoagulant compounds
regarding efficacy but more importantly safety. In the BT experiments the baseline
results were compatible with the previously reported data, and intravenously administered
APACs- and UFH-modified hemostasis as shown by the prolongation of the aPTT and BT.[25]
[26] APAC1 and APAC2 induced 50% less bleeding and prolonged aPTT at least 15% less in
comparison with UFH at the similar dose. APAC1 showed dose-dependent effects on prolonging
the BT at the selected dose range while APAC2 with the doubled coupling level of heparin
chains induced less bleeding. APACs may limit simultaneously platelet function by
inhibiting collagen-induced platelet aggregation and modifying the intrinsic coagulation
pathway and anticoagulants in vivo. Importantly, no significant changes were detected
in platelet number or other blood cell count during and after these experiments.
The two in vivo experiments in baboons are encouraging regarding the dual antiplatelet
and anticoagulant action.[27]
[28] In the modified Folt model of acute thrombosis in baboons, APAC applied locally
on the crush-injured and stenosed arterial site effectively and consistently inhibited
thrombosis in the artery under the moderate shear stress, with the flow rate of 100
mL/min and stenosis of 30% until the experiment was interrupted after 120 minutes.
When APAC was addressed with the short exposure of sequentially increased stenosis
from 60 to 90% the patency of the artery was further maintained. In addition, in the
extracorporeal collagen graft model, APAC inhibited both thrombosis and fibrin accumulation
about by 50% over UFH in the 60-minute experiment in baboons. The APAC potency was
further emphasized with the distally propagated thrombus over the effect of heparin.
APAC has potential to target itself at the vascular injury site. The local administration
and residence of APAC on the femoral artery anastomosis was confirmed with the PET
scan in rats where the retention time of copper-labeled APAC was at least twice the
time of UFH. This finding refers to the capacity of APACs to target at the injury
site and to be maintained there for a significant period of time to inhibit unwanted
thrombotic response. As the vascular injury triggers the inflammatory cellular triad
of the leukocytes, lymphocytes, and mast cells with their heparin proteoglycans released
to the tissue, the local heparin reaction is a native physiological defense mechanism.
Overall, APAC dually inhibited collagen- and ristocetin-induced platelet aggregation
in human blood, with anticoagulant properties that were at least comparable with UFH
in plasma. In the absence of systemic antithrombotic therapy, treatment of a highly
thrombogenic vascular and collagen surfaces with APAC arrested in vivo thrombus formation
in two models of acute thrombosis in baboons. Locally administered APAC alone, with
its dual antiplatelet and anticoagulant effects, may limit the growth of thrombus
and prevent thrombo-occlusions in various applications. The encouraging data in the
BT model would support even the systemic administration of APAC. However, the local
application and administration to thrombotic sites of vascular injury would represent
a novel modality to treat arterial thrombosis in association with interventions and
risk of acute thrombosis. This model of antithrombotic tissue action would be compatible
with nature's GAGs, including large heparin conjugates derived from mast cells.




