

Subscribe to RSS
DOI: 10.1055/s-0034-1379637
Stereoselective Suzuki Coupling Reaction of an α-Bromo-α-fluoro-β-lactam
Authors
Publication History
Received: 22 September 2014
Accepted after revision: 09 November 2014
Publication Date:
28 November 2014 (online)

Abstract
A new strategy has been developed for the synthesis of α-aryl-α-fluoro-β-lactams via the Suzuki cross-coupling of α-bromo-α-fluoro-β-lactam with a range of different aryl-(9-BBN) reagents. This method provides facile access to multisubstituted α-fluoro-β-lactams in a diastereoselective manner. The synthetic utility of α-bromo-α-fluoro-β-lactam has been demonstrated by the arylation of α-bromo-α-fluoro-β-lactam.
Organofluorine compounds are becoming increasingly popular with a growing number of applications in a variety of fields, including medicine, agrochemicals and materials science.[1] The introduction of a fluorine substituent can have a dramatic effect on the dipole moment of the parent compound and enhance its electrophilicity, which can be an attractive property in the drug discovery and medicinal chemistry investigations.[2]

In terms of potential strategies for the introduction of fluorine, site-selective approaches involving the fluorination of a target molecule are particularly desirable, because they can potentially allow for the late-state introduction of fluorine substituents. Compared with the direct fluorination of the parent molecule, the use of fluorine-containing building blocks represents one of the most efficient and practical methods for the site-selective introduction of a fluorine substituent. β-Lactams are a well-known structural class of bioactive compounds, including antibiotics and hypercholesterol-lowering agents.[3] The incorporation of a fluorine atom into a β-lactam ring can be beneficial in terms of the enhancing the electrophilicity of the parent compound, which can have a dramatic effect on the potency and drug metabolism and pharmacokinetics (DMPK) properties of the compound.[4] Furthermore, fluorinated β-lactams have been used as building blocks for the construction of fluorine-containing β-amino acid units,[5] which could be used to prevent epimerization at the α position of the β-amino acid backbone. We recently reported the synthesis of several non-chiral and chiral fluorinated β-lactams,[6] [7] and have also demonstrated the utility of α-bromo-α-fluoro-β-lactam (1) using a nickel-catalyzed cross-coupling reaction and lithiation chemistry to provide access to multisubstituted α-fluoro-β-lactams (Equation 1).[8] Although our reported Kumada coupling reaction was useful for the arylation of 1,[8a] the reaction did not afford any of the ortho-substituted arylated products. The nickel-catalyzed Negishi and Suzuki coupling reactions of fluorinated halo substrates have been reported by Fu et al.[9] and Gandelman et al.[10] to provide the corresponding arylated α-fluoro carbonyl compounds and secondary alkyl fluorides, respectively. Fu et al. also reported a similar reaction for the nickel-catalyzed Suzuki arylation of alkyl halide using aryl-(9-BBN) reagents.[11] To the best of our knowledge, however, there have been no reports in the literature concerning the direct functionalization of fluoro-β-lactams, except those of our own group and Welch’s group.[12] To develop a new approach for the construction of multisubstituted α-fluoro-β-lactams, we investigated the cross-coupling reactions of 1. In this study, we describe the Suzuki cross-coupling reaction of 1 with aryl-(9-BBN) reagents, which allowed for the synthesis of multisubstituted α-fluoro-β-lactams with high diastereoselectivity (Scheme [1]). In a further demonstration of the synthetic utility of this strategy, we have also completed the synthesis of the highly enantiomerically enriched α-arylated product 3 starting from chiral 1.

Several reaction conditions were initially investigated for the Suzuki coupling reaction of the model substrate 1-benzyl-3-bromo-3-fluoro-4-phenylazetidin-2-one (1a) with phenyl-(9-BBN) (2a). When 1a was reacted with 2a using tert-BuOLi as a base, NiCl2·DME as a catalyst and bipyridine (L1; Figure [1]) as a ligand, the desired arylated product (3a) was formed diastereoselectively, albeit in a low yield (Table [1], entry 1).[13] Several other tert-butoxides salts were also investigated, including sodium and potassium salts, but these salts gave none of the desired coupling product (Table [1], entries 2 and 3).
a Isolated yield.
b 19F NMR yield.
c Amount of Ni catalyst used was 20 mol%.
d Amount of ligand was used was 22 mol%.


The poor results obtained with these salts were attributed to the poor solubility profiles of sodium and potassium tert-butoxides in organic solvents. Interestingly, the use of NiBr2·diglyme instead of NiCl2·DME led to a slight increase in the yield (Table [1], entry 4). Several other bipyridyl ligands were also screened in the reaction, but resulted in the recovery of 1a and low yields of the desired product (Table [1], entries 4–7). Pleasingly, the use of 4,4′-di-tert-butyl-2,2′-bipyridine (L5; Figure [1]) as a ligand in this Suzuki coupling reaction led to the highest yield of 3a, with all of the starting material 1a being consumed (Table [1], entry 8). Further attempts to optimize the reaction conditions, including changing the solvent, led to much lower yields of the product especially, when the reaction was carried out in THF or toluene (Table [1], entries 9–12). Increasing the amount of catalyst and ligand added to the reaction led to an increase in the yield of 3a (Table [1], entry 13). A further improvement in the product yield was also observed when the reaction was conducted under reflux conditions, which also led to a significant reduction in the reaction time (Table [1], entry 14). Several other boron reagents were also investigated in this coupling reaction, but these reagents failed to provide any of the desired products, with the starting material being recovered in both cases (Table [1], entries 15 and 16). It is noteworthy that the coupling product 3a was obtained as a single diastereomer in all cases.[14] With the optimized conditions in hand, we proceeded to evaluate the scope of the Suzuki coupling reaction of 1a with a variety of aryl-(9-BBN) reagents, which gave the corresponding multisubstituted fluoro-β-lactams in good yields (Scheme [2]). Thus, para- and meta-substituted aryl-(9-BBN) reagents performed well in the cross-coupling reaction to give the desired α-arylated products in good yields with high levels of diastereoselectivity. Notably, both electron-rich and electron-poor nucleophiles performed well as the coupling partners (e.g., 2b–h). The ortho-substituted aryl-(9-BBN) reagents performed especially well in the reaction, with the desired products 3i and 3j being formed in good yields. Furthermore, the heterocyclic aryl-(9-BBN) reagent 2k reacted successfully under the optimized conditions to give the corresponding product 3k in moderate yield. In contrast, alkyl-(9-BBN) and alkenyl-(9-BBN) reagents were found to be unsuitable coupling partners for the reaction.
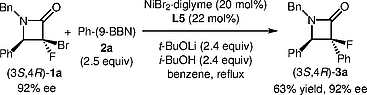
The α-aryl-α-fluoro-β-lactams 3 described in the current study were all prepared as racemic mixtures. To expand the scope of this chemistry, we also investigated the asymmetric synthesis of these multisubstituted α-fluoro-β-lactams using chiral 1a with phenyl-(9-BBN) (Scheme [3]). Pleasingly, the Suzuki coupling reaction of chiral 1a in the 3S,4R-configuration (92% ee) provided the enantiomerically enriched product 3a in a diastereoselective manner without any decrease in its enantiopurity (63% yield, 92% ee).
In summary, we have developed a new reaction for highly diastereoselective construction of α-arylated α-fluoro-β-lactams via the nickel-catalyzed Suzuki coupling reaction of α-bromo-α-fluoro-β-lactam with aryl-(9-BBN) reagents. The chiral coupling product was also obtained diastereoselectively without any reduction in its enantiopurity when the reaction was conducted with enantioenriched α-bromo-α-fluoro-β-lactam. Additional investigations towards the synthesis of multisubstituted α-fluoro-β-lactams are currently underway in our laboratory.
Supporting Information
- Supporting information for this article is available online at http://dx.doi.org/10.1055/s-0034-1379637.
- Supporting Information (PDF) (opens in new window)
-
References and Notes
- 1a Jeschke P. ChemBioChem 2004; 5: 570
- 1b Müller K, Faeh C, Diederich F. Science 2007; 317: 1881
- 1c Zhang W, Cai C. Chem. Commun. 2008; 5686
- 1d O’Hagan D. J. Fluorine Chem. 2010; 131: 1071
- 2 Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008; 37: 320
- 3a Rosenblum SB, Huynh T, Afonso A, Davis HR, Yumibe N, Clader JW, Burnett DA. J. Med. Chem. 1998; 41: 973
- 3b Kværnø L, Werder M, Hauser H, Carreira EM. J. Med. Chem. 2005; 48: 6035
- 4 Hagmann WK. J. Med. Chem. 2008; 51: 4359
- 5a Uoto K, Ohsuki S, Takenoshita H, Ishiyama T, Iimura S, Hirota Y, Mitsui I, Terasawa H, Soga T. Chem. Pharm. Bull. 1997; 45: 1793
- 5b Li X.-G, Lähitie M, Kanerva LT. Tetrahedron: Asymmetry 2008; 19: 1857
- 5c Liu N, Cao S, Shen L, Wu J, Yu J, Zhang J, Li H, Qian X. Tetrahedron Lett. 2009; 50: 1982
- 6a Sato K, Tarui A, Matsuda S, Omote M, Ando A, Kumadaki I. Tetrahedron Lett. 2005; 46: 7679
- 6b Tarui A, Kawashima N, Sato K, Omote M, Miwa Y, Minami H, Ando A. Tetrahedron Lett. 2010; 51: 2000
- 7a Tarui A, Nishimura H, Ikebata T, Tahira A, Sato K, Omote M, Minami H, Miwa Y, Ando A. Org. Lett. 2014; 16: 2080
- 7b Tarui A, Ikebata T, Sato K, Omote M, Ando A. Org. Biomol. Chem. 2014; 12: 6484
- 8a Tarui A, Kondo S, Sato K, Omote M, Minami H, Miwa Y, Ando A. Tetrahedron 2013; 69: 1559
- 8b Tarui A, Kawashima N, Kawakita T, Sato K, Omote M, Ando A. J. Org. Chem. 2013; 78: 7903
- 9 Liang Y, Fu GC. J. Am. Chem. Soc. 2014; 136: 5520
- 10 Jiang X, Sakthivel S, Kulbitski K, Nisnevich G, Gandelman M. J. Am. Chem. Soc. 2014; 136: 9548
- 11a Lundin PM, Fu GC. J. Am. Chem. Soc. 2010; 132: 11027
- 11b Wilsily A, Tramutola F, Owston NA, Fu GC. J. Am. Chem. Soc. 2012; 134: 5794
- 11c Zultanski SL, Fu GC. J. Am. Chem. Soc. 2013; 135: 624
- 12a Welch JT, Araki K, Kawecki R, Wichtowski JA. J. Org. Chem. 1993; 58: 2454
- 12b Kawecki R, Welch JT. Tetrahedron Lett. 1993; 34: 3087
- 13 Typical Experimental Procedure for the Ni-Catalyzed Suzuki Coupling Reaction of α-Bromo-α-fluoro-β-lactam: 4,4′-Di-tert-butylbipyridine (29.5 mg, 0.11 mmol) and NiBr2·diglyme (35 mg, 0.10 mmol) were added to a flask equipped with a magnetic stirrer bar. To the flask was added anhyd benzene (7.5 mL) and the resulting mixture was stirred vigorously for 2 h (a light-green slurry formed). The solution of the activated Ph-(9-BBN) solution (1.25 mmol) was added to the slurry, and the whole mixture was stirred for 20 min at same temperature. Then, 1 (0.5 mmol) was added to the slurry and the resulting mixture was stirred for 1 h under reflux. The reaction was quenched by brine and the mixture was extracted with EtOAc, and then the extract was dried over MgSO4. The solvent was removed in vacuo and the residue was purified by silica gel column chromatography (hexane–EtOAc) to give the desired product 3.(3S,4R/3R,4S)-1-Benzyl-3-fluoro-3,4-diphenylazetidin-2-one (3a): colorless solid (145 mg, 87%); mp 78.0–79.0 °C (uncorrected). 1H NMR (400 MHz, CDCl3): δ = 3.98 (dd, J = 14.8, 2.4 Hz, 1 H), 4.66 (d, J = 3.5 Hz, 1 H), 5.03 (d, J = 14.8 Hz, 1 H), 7.16–7.18 (m, 2 H), 7.29–7.33 (m, 5 H), 7.38 (m, 5 H), 7.42–7.44 (m, 3 H). 13C NMR (100 MHz, CDCl3): δ = 44.4 (d, J = 2 Hz), 68.6 (d, J = 25 Hz), 102.2 (d, J = 225 Hz), 125.2 (d, J = 7 Hz), 128.0, 128.2 (d, J = 2 Hz), 128.5, 128.6, 128.7, 128.8, 129.0, 129.2 (d, J = 2 Hz), 132.1, 134.2 (d, J = 24 Hz), 134.4, 164.8 (d, J = 25 Hz). 19F NMR (84 MHz, CDCl3): δ = –102.5 (s, 1 F). MS: m/z = 331 [M+]. HRMS (EI): m/z [M+] calcd for C22H18FNO: 331.1372; found: 331.1378.
- 14 See supporting information for the determination of the relative configuration and the diastereoselective formation of coupling products.
- 15 The ee value was determined to be 92% by HPLC analysis [Daicel CHIRALPAK AD-H, hexane–EtOH = 98:2, flow rate = 2.0 mL/min, λ = 254 nm, t R (major) = 10.0 min and t R (minor) = 8.9 min]. [α]D 25 +63.4 (c = 1.05, CHCl3). See the Supporting Information for enantiopurity of 3a.
-
References and Notes
- 1a Jeschke P. ChemBioChem 2004; 5: 570
- 1b Müller K, Faeh C, Diederich F. Science 2007; 317: 1881
- 1c Zhang W, Cai C. Chem. Commun. 2008; 5686
- 1d O’Hagan D. J. Fluorine Chem. 2010; 131: 1071
- 2 Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008; 37: 320
- 3a Rosenblum SB, Huynh T, Afonso A, Davis HR, Yumibe N, Clader JW, Burnett DA. J. Med. Chem. 1998; 41: 973
- 3b Kværnø L, Werder M, Hauser H, Carreira EM. J. Med. Chem. 2005; 48: 6035
- 4 Hagmann WK. J. Med. Chem. 2008; 51: 4359
- 5a Uoto K, Ohsuki S, Takenoshita H, Ishiyama T, Iimura S, Hirota Y, Mitsui I, Terasawa H, Soga T. Chem. Pharm. Bull. 1997; 45: 1793
- 5b Li X.-G, Lähitie M, Kanerva LT. Tetrahedron: Asymmetry 2008; 19: 1857
- 5c Liu N, Cao S, Shen L, Wu J, Yu J, Zhang J, Li H, Qian X. Tetrahedron Lett. 2009; 50: 1982
- 6a Sato K, Tarui A, Matsuda S, Omote M, Ando A, Kumadaki I. Tetrahedron Lett. 2005; 46: 7679
- 6b Tarui A, Kawashima N, Sato K, Omote M, Miwa Y, Minami H, Ando A. Tetrahedron Lett. 2010; 51: 2000
- 7a Tarui A, Nishimura H, Ikebata T, Tahira A, Sato K, Omote M, Minami H, Miwa Y, Ando A. Org. Lett. 2014; 16: 2080
- 7b Tarui A, Ikebata T, Sato K, Omote M, Ando A. Org. Biomol. Chem. 2014; 12: 6484
- 8a Tarui A, Kondo S, Sato K, Omote M, Minami H, Miwa Y, Ando A. Tetrahedron 2013; 69: 1559
- 8b Tarui A, Kawashima N, Kawakita T, Sato K, Omote M, Ando A. J. Org. Chem. 2013; 78: 7903
- 9 Liang Y, Fu GC. J. Am. Chem. Soc. 2014; 136: 5520
- 10 Jiang X, Sakthivel S, Kulbitski K, Nisnevich G, Gandelman M. J. Am. Chem. Soc. 2014; 136: 9548
- 11a Lundin PM, Fu GC. J. Am. Chem. Soc. 2010; 132: 11027
- 11b Wilsily A, Tramutola F, Owston NA, Fu GC. J. Am. Chem. Soc. 2012; 134: 5794
- 11c Zultanski SL, Fu GC. J. Am. Chem. Soc. 2013; 135: 624
- 12a Welch JT, Araki K, Kawecki R, Wichtowski JA. J. Org. Chem. 1993; 58: 2454
- 12b Kawecki R, Welch JT. Tetrahedron Lett. 1993; 34: 3087
- 13 Typical Experimental Procedure for the Ni-Catalyzed Suzuki Coupling Reaction of α-Bromo-α-fluoro-β-lactam: 4,4′-Di-tert-butylbipyridine (29.5 mg, 0.11 mmol) and NiBr2·diglyme (35 mg, 0.10 mmol) were added to a flask equipped with a magnetic stirrer bar. To the flask was added anhyd benzene (7.5 mL) and the resulting mixture was stirred vigorously for 2 h (a light-green slurry formed). The solution of the activated Ph-(9-BBN) solution (1.25 mmol) was added to the slurry, and the whole mixture was stirred for 20 min at same temperature. Then, 1 (0.5 mmol) was added to the slurry and the resulting mixture was stirred for 1 h under reflux. The reaction was quenched by brine and the mixture was extracted with EtOAc, and then the extract was dried over MgSO4. The solvent was removed in vacuo and the residue was purified by silica gel column chromatography (hexane–EtOAc) to give the desired product 3.(3S,4R/3R,4S)-1-Benzyl-3-fluoro-3,4-diphenylazetidin-2-one (3a): colorless solid (145 mg, 87%); mp 78.0–79.0 °C (uncorrected). 1H NMR (400 MHz, CDCl3): δ = 3.98 (dd, J = 14.8, 2.4 Hz, 1 H), 4.66 (d, J = 3.5 Hz, 1 H), 5.03 (d, J = 14.8 Hz, 1 H), 7.16–7.18 (m, 2 H), 7.29–7.33 (m, 5 H), 7.38 (m, 5 H), 7.42–7.44 (m, 3 H). 13C NMR (100 MHz, CDCl3): δ = 44.4 (d, J = 2 Hz), 68.6 (d, J = 25 Hz), 102.2 (d, J = 225 Hz), 125.2 (d, J = 7 Hz), 128.0, 128.2 (d, J = 2 Hz), 128.5, 128.6, 128.7, 128.8, 129.0, 129.2 (d, J = 2 Hz), 132.1, 134.2 (d, J = 24 Hz), 134.4, 164.8 (d, J = 25 Hz). 19F NMR (84 MHz, CDCl3): δ = –102.5 (s, 1 F). MS: m/z = 331 [M+]. HRMS (EI): m/z [M+] calcd for C22H18FNO: 331.1372; found: 331.1378.
- 14 See supporting information for the determination of the relative configuration and the diastereoselective formation of coupling products.
- 15 The ee value was determined to be 92% by HPLC analysis [Daicel CHIRALPAK AD-H, hexane–EtOH = 98:2, flow rate = 2.0 mL/min, λ = 254 nm, t R (major) = 10.0 min and t R (minor) = 8.9 min]. [α]D 25 +63.4 (c = 1.05, CHCl3). See the Supporting Information for enantiopurity of 3a.
