Keywords
NETs - cancer - thrombosis - biomarkers
The scientific and medical community has in recent years increased its attention to
neutrophils. New important functions and properties of these immune cells are emerging
in addition to their role as the first line of defense against pathogens. The discoveries
surrounding neutrophils promoted them from simple phagocytes to a sophisticated population
of innate immune cells which orchestrate both the innate and adaptive inflammatory
response.[1]
[2] Among these novel characteristics, the formation of neutrophil extracellular DNA
traps (NETs) has defined new roles for neutrophils in inflammation and immunity but
also in pathological conditions including cancer biology and thrombosis.
Neutrophil Extracellular Traps
Neutrophil Extracellular Traps
The first evidence that neutrophils release their chromatin in the extracellular space
was provided in 2004 by Arturo Zychlinsky's group.[3] The authors showed that upon activation, neutrophils generate extracellular DNA
fibers composed of granular proteases and nuclear constituents that trap and kill
bacteria, thus defining a new form of antimicrobial innate response. Further in vitro
characterizations described NETosis as a novel cell death program leading to the decondensation
of the chromatin followed by nuclear and granular membrane disintegration, mixing
of the components, and finally cytoplasmic membrane lysis and NETs release.[4] In some cases, a more rapid ejection of the chromatin through vesicular exocytosis
of nuclear contents was observed.[5] In vivo, intact anuclear neutrophils that released only their nuclear contents without
lysis were also observed. These “multitasking” cells retained the ability to crawl
and phagocytose microbes in vivo.[6] Recently, the term “vital NETosis” was proposed to distinguish it from NETosis by
cell lysis[4] and it has been suggested that different subsets of neutrophils or different stimuli
could lead to one or the other.[7]
The molecular mechanisms leading to NET formation remain unclear. The release of the
chromatin by neutrophils was originally shown to be dependent on the generation of
reactive oxygen species (ROS).[4] The production of ROS occurs through the activation of nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase, RAF-MEK-ERK, and p38MAPK pathways[8]
[9]
[10] whereas other stimuli activate NETosis in an NADPH oxidase independent manner.[5]
[11] It has also been suggested that neutrophil elastase and myeloperoxidase are implicated
in the process.[12] Upon activation, neutrophil elastase translocates to the nucleus and helps chromatin
decondensation by partially degrading histones. The chromatin decondensation is primarily
driven by another modification of histones, histone citrullination through the activation
of peptidylarginine deiminase 4 (PAD4). PAD4 (also called PADI4) is an enzyme highly
expressed in many cancers[13] and in neutrophils where it is crucial to NET formation.[14]
[15]
[16] The critical importance of PAD4 and histone citrullination has been documented by
the use of PAD4 inhibition and treatment with a PAD4 fusion protein in HL-60 cells[16] as well as by genetically engineered PAD4-deficient mice whose neutrophils are unable
to form NETs.[14] Moreover, PAD4 overexpression in an osteosarcoma cell line was sufficient to induce
chromatin decondensation and release, proving its function in this artificial setting.[17] How PAD4 expression and activity are regulated in neutrophils and why NETosis sometimes
results in only nuclear release and other times in cell lysis remain to be elucidated.
Neutrophil Extracellular Traps in Thrombosis
Neutrophil Extracellular Traps in Thrombosis
The ejection of neutrophil chromatin decorated with granular proteins and proteases
at sites of infection traps and kills pathogens, conferring a protection to the host.
However, the release of chromatin in the vasculature of an uninfected animal can be
quite deleterious. Indeed, NETs promote thrombosis by providing a scaffold for platelet
and red blood cell adhesion and aggregation[18] and enhancing coagulation.[19] Biomarkers of NETs have been observed in thrombi and plasma of baboons and mice
subjected to deep vein thrombosis (DVT), revealing NETs as a structural part of the
thrombus.[18]
[20] The presence of NETs has also been seen in a human thrombus[21] and in plasma of patients with DVT.[22]
[23] It was suggested that thrombus neutrophils, through platelet-dependent stimulation,
were indispensable for the activation of factor XII and propagation of the thrombus
in the inferior vena cava stenosis model of DVT in mice.[24] Importantly, using PAD4-deficient mice, NETs were shown to be not only part of venous
thrombi but to be critical to their formation and/or persistence.[15] Thrombosis in PAD4-deficient mice can be rescued by wild-type neutrophil infusion
showing their importance and sufficiency in the pathogenesis of DVT in mice.[15] In humans, plasma markers of NETs correlate with thrombotic diseases activity such
as thrombotic microangiopathies (TMA)[25] and also coronary atherothrombosis.[26]
Exactly how NETs promote coagulation and thrombosis may depend on the circumstances
in which they form. The major constituents of NETs, DNA, histones, and proteases,
all have procoagulant properties. In vitro, extracellular nucleic acids, including
genomic DNA, enhance protease activity of coagulation factors,[27] and induce thrombin generation in platelet-poor plasma.[28] In addition, histones are cytotoxic to the endothelium and can induce macro and
microthrombosis in vivo.[29] Histones inhibit anticoagulation of plasma by impairing thrombomodulin function,[30] and promoting thrombin generation[31] and platelet aggregation resulting in thrombocytopenia.[32] Serine proteases, such as neutrophil elastase, have also been shown to inactivate
tissue factor pathway inhibitor thus further increasing coagulation and fibrin deposition
in vivo.[19] Tissue factor has also been observed on NETs.[33]
[34] Thus in many ways, the release of NETs in the vascular compartment triggers a procoagulant
state and promotes binding and activation of platelets leading to thrombosis.
Cancer and NETosis
Neutrophils play a major role in cancer biology.[35] They make up a significant portion of the inflammatory cell infiltrate in many mouse
models of cancer and also in human tumors.[36] However, their role in tumor progression is still debated because both pro- and
antitumoral properties have been attributed to neutrophils.[35]
[36] It was suggested that the opposing roles of tumor-associated neutrophils could be
related to their stage of activation.[36] Thus, NETs, which may be produced only by a subset of neutrophils, may potentially
contribute to one of these properties. The release of the neutrophil's chromatin may
influence many different steps of tumor development including tumor growth, angiogenesis,
metastasis, and immune suppression.[37] Our group reported the presence of a large area of dead neutrophils and NET-like
structures in Lewis lung carcinoma (LLC) hemorrhagic tumors.[38] A more detailed analysis of the LLC tumors by confocal microscopy clearly revealed
the presence of large areas of neutrophil accumulation within the tumors ([Fig. 1A]). These apparently necrotic areas are filled with not only intact neutrophils but
also primed hypercitrullinated neutrophils ready to generate NETs and extracellular
chromatin containing hypercitrullinated histones. A similar pattern was observed in
histological sections of tumors of two patients with Ewing sarcoma.[39] Thus, NETs are found in tumors at sites of neutrophil accumulation and may influence
the cancer microenvironment. Whether neutrophils are recruited and NETs formed because
of the inflammatory/hypoxic environment of the tumor or NETs are responsible for the
generation of necrotic areas remains to be clarified. Hypoxia within the tumor, for
example, may activate hypoxia-inducible factor 1α, which in turn may stimulate NETosis.[40] Intriguingly, a recent study demonstrated that the phenotype of the tumor neutrophils
is dependent on the tumor stage, becoming more tumor promoting as the tumor ages.[41] The study shows that at an early stage of tumor development, the antitumoral neutrophils
are localized around the tumor whereas at a later time point the protumoral neutrophils
accumulate inside the tumor. As NETs are observed within the tumor, this suggests
an advantageous effect of NETs on primary tumor growth.
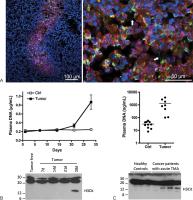 Fig. 1 Presence of neutrophil extracellular traps (NETs) in the tumor and in the plasma
of tumor-bearing hosts. (A) Intratumoral presence of NETs. Lewis lung carcinoma tumor
cells were injected subcutaneously in the right flank of C57BL/6 mice and tumors allowed
to grow. At day 17, mice were euthanized and tumors collected and snap-frozen. Forty
micrometers sections were stained with anti-Ly6G (red, a neutrophil marker), anti-H3Cit
(green) and DNA counterstained with Hoechst 33342 (blue). Confocal microscopy images
were taken at least 5 µm deep into the section to avoid artifacts. Low magnification
representative image (left) shows a large area rich in neutrophils at the center of
the tumor. H3Cit-positive neutrophils and extracellular chromatin are observed. High
magnification image from this area (right) shows the presence of H3Cit-positive neutrophils
but also extracellular H3Cit-containing strings of DNA (arrows). (B, C) High levels
of plasma DNA and NET biomarkers are found in murine and human thrombotic hosts with
cancer. (B) High levels of plasma DNA and H3Cit were found in mice with 4T1 breast
cancer at a late stage of the disease. Reprinted with permission from Demers et al.[58] (C). High level of plasma DNA and the presence of H3Cit were also observed in most
of the cancer patients with acute thrombocytic microangiopathies (TMA). The DNA results
were adapted from Fuchs et al.[25] This research was originally published in the journal Blood (Fuchs TA, Kremer Hovinga
JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate
disease activity in patients with thrombotic microangiopathies. Blood 2012;120(6):1157–1164.
© American Society of Hematology). The method for H3Cit analysis is described in Demers
et al.[58]
Fig. 1 Presence of neutrophil extracellular traps (NETs) in the tumor and in the plasma
of tumor-bearing hosts. (A) Intratumoral presence of NETs. Lewis lung carcinoma tumor
cells were injected subcutaneously in the right flank of C57BL/6 mice and tumors allowed
to grow. At day 17, mice were euthanized and tumors collected and snap-frozen. Forty
micrometers sections were stained with anti-Ly6G (red, a neutrophil marker), anti-H3Cit
(green) and DNA counterstained with Hoechst 33342 (blue). Confocal microscopy images
were taken at least 5 µm deep into the section to avoid artifacts. Low magnification
representative image (left) shows a large area rich in neutrophils at the center of
the tumor. H3Cit-positive neutrophils and extracellular chromatin are observed. High
magnification image from this area (right) shows the presence of H3Cit-positive neutrophils
but also extracellular H3Cit-containing strings of DNA (arrows). (B, C) High levels
of plasma DNA and NET biomarkers are found in murine and human thrombotic hosts with
cancer. (B) High levels of plasma DNA and H3Cit were found in mice with 4T1 breast
cancer at a late stage of the disease. Reprinted with permission from Demers et al.[58] (C). High level of plasma DNA and the presence of H3Cit were also observed in most
of the cancer patients with acute thrombocytic microangiopathies (TMA). The DNA results
were adapted from Fuchs et al.[25] This research was originally published in the journal Blood (Fuchs TA, Kremer Hovinga
JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate
disease activity in patients with thrombotic microangiopathies. Blood 2012;120(6):1157–1164.
© American Society of Hematology). The method for H3Cit analysis is described in Demers
et al.[58]
The involvement of NETs in promoting metastasis has recently been elegantly shown
using intravital microscopy.[42] In a model of systemic infection, a condition promoting metastasis, the authors
documented NETs deposition on the microvasculature and subsequent trapping of circulating
cancer cells in the DNA web. It was known that, during septicemia, NETs are released
in the vasculature[43] where they trap the bacteria.[44] The authors hypothesized that, similar to the immobilization of bacteria, NETs would
immobilize the tumor cells. Indeed, they show that NETs-entrapped tumor cells survive
and proliferate to form nodules.[42] It is possible that NETs or their degradation products are inhibiting immune cells
allowing better survival. The observed increase in tumor metastasis again suggests
a role for NETs in enhancing tumor progression. The effect of NETs could be attenuated
by treatment with DNase, an enzyme that cleaves the DNA backbone of NETs.[3] Thus, the presence of intravascular NETs following sepsis promotes metastasis in
mice. Whether NETs just protect or anchor cancer cells physically or whether they
generate thrombin that promotes tumor growth[45]
[46]
[47]
[48]
[49] remains to be determined.
Neutrophil Extracellular Traps in Cancer-Associated Thrombosis
Neutrophil Extracellular Traps in Cancer-Associated Thrombosis
The association of cancer and thrombosis was reported in the 19th century by Armand
Trousseau, describing thrombosis as a presenting feature even before the diagnosis
of cancer; this is now defined as Trousseau syndrome.[50] Over the years, studies have shown that tissue factor, microparticles, cytokines,
soluble P-selectin, elevation in coagulation factors, secretion of mucins, thrombocytosis,
and leukocytosis are implicated in the prothrombotic state associated with cancers.[51]
[52]
[53] Interestingly, in 1986, Shoenfeld et al observed that the leukocytosis in 10 different
types of nonhematologic malignancies was attributed mainly to an increase in polymorphonuclear
cells and was associated with a poor clinical outcome.[54] Indeed, recently, an increase in peripheral neutrophil counts and/or intratumoral
neutrophils or a high neutrophil to lymphocyte ratio were all linked to poor prognosis
and outcome in many different types of cancer.[55] In humans, this elevation in neutrophils was seen mainly in renal cell carcinoma,
melanoma, hepatocellular carcinoma, glioblastoma, colorectal, gastric, esophageal,
lung, ovarian, and head and neck cancer, most of which are associated with a high
risk of venous thromboembolism.[56]
Using a murine model of mammary carcinoma, in which a leukemoid reaction leading to
an increase in blood neutrophil has been described,[57] we recently showed occurrence of spontaneous thrombosis.[58] Lung thrombosis was observed at a late stage of the disease when the neutrophil
count and level of plasma DNA were extremely high ([Fig. 1B]). Surprisingly, when neutrophils from tumor-bearing mouse blood were analyzed, an
increasing number of highly hypercitrullinated neutrophils, ready to generate NETs,
were observed as the tumor progressed. At the late time point where thrombosis was
identified, a reduced number of highly hypercitrullinated neutrophils were counted
and hypercitrullination of histone H3 (H3Cit) was detected in the plasma ([Fig. 1B]), suggesting that NETosis was spontaneously occurring.[58] The generation of NETs and the associated prothrombotic state can also be induced
in tumor-bearing mice at an early time point of tumor progression by injection of
low-dose lipopolysaccharides (LPS).
The predisposition of tumor-induced neutrophils to NET formation was confirmed in
vitro in mammary and lung carcinoma as well as chronic myelogenous leukemia.[58] The molecular mechanism leading to the priming of tumor-induced neutrophils seems
to be related to an increase in plasma granulocyte colony-stimulating factor (G-CSF).
Indeed, the elevation in neutrophil count, neutrophil priming, and the prothrombotic
phenotype could be mimicked by administration of G-CSF before LPS in tumor-free mice.
G-CSF-producing tumors have also been associated with poor prognosis.[59]
[60]
[61]
The thrombotic state produced upon NET generation seems to be related to the DNA scaffold.
Digestion of NETs by pretreatment with DNase before LPS injection prevented thrombosis
in tumor-bearing mice.[58] High levels of plasma DNA in cancer patients were described more than 30 years ago.[62] Since then, an increase in plasma or serum DNA has been observed in many different
types of cancer.[63] The DNA was long thought to be released either from the tumor or the host-injured
tissue by apoptotic or necrotic cell death.[64] In fact, it has been shown that a large amount of nontumoral DNA is found in the
plasma during tumor progression in rat models.[65]
[66] High plasma DNA has also been observed in cases of inflammatory disease and trauma.[63] Thus, the identification of NETs in cancer, as well as in other inflammatory diseases,
now provides another explanation for the elevated plasma DNA. The association of plasma
DNA with neutrophil counts and NETs biomarkers, such as H3Cit, could be used as diagnostic
tools to evaluate propensity to thrombosis. Indeed, patients with tumor-associated
TMA show high levels of plasma DNA ([Fig. 1C]) that are associated with NETs markers myeloperoxidase and calprotectin.[25] Moreover, on further analysis, we now show that H3Cit can be detected in the stored
plasma from most of these cancer patients with an acute episode of TMA whereas no
H3Cit was found in the plasma of healthy controls ([Fig. 1C]). Thus, cell-free H3Cit can be observed in the plasma of cancer patients with thrombotic
complications, just as it was observed in mice.[58] This emphasizes H3Cit's potential as a biomarker for cancer-associated thrombosis.
The contribution of NETs to the total level of plasma DNA found in cancer hosts remain
to be addressed.
In our mouse models, tumor-induced neutrophils are sensitized to NET formation and
can easily release their chromatin and promote thrombosis upon encountering a second
hit even at an early stage of the disease. Thus, what happens after chemotherapy?
It is known that chemotherapies induce cell death and release of DNA in the plasma
and are associated with a high risk of thrombosis.[67] Large DNA fragments are found in the plasma of patients after the first cycle of
docetaxel chemotherapy for prostate cancer[68] and neoadjuvant chemotherapy for breast cancer.[69] The appearance of these large DNA fragments was attributed to rapid tumor cell necrosis
following treatment. With our new knowledge on NETs, it is plausible that chemotherapies
could either directly induce NETosis or serve as the second hit in NET generation.
Indeed, Swystun et al reported that doxorubicin, epirubicin, and 5-fluororuacil, three
breast cancer therapeutic agents, induce the release of DNA and thrombin–antithrombin
complexes (TAT) when injected in healthy mice.[28] However, in vitro, only doxorubicin and epirubicin had the potential to induce DNA
release from isolated neutrophils suggesting that some drugs are indeed sufficient
for NET induction. In vitro, in venous whole blood, the release of DNA could be prevented
by the addition of the antioxidant glutathione, which prevents damage by ROS and thus
NETosis. Moreover, the authors showed that the cell-free DNA released induces thrombin
generation in whole blood, and that an increase in cell-free DNA and TAT are found
in the plasma of early-stage breast cancer patients 24 hours postchemotherapy,[28] a time point where thrombotic events are likely to occur.[70] Thus, this study defined NETs as a novel procoagulant linking chemotherapy to thrombosis.[70]
Conclusion
The discovery of the formation of NETs by stimulated neutrophils unveiled unexpected
functions of neutrophils in inflammatory diseases, cancer, and thrombosis. Many aspects
of this new field still need to be addressed. Through the release of chromatin in
tissues or in the vasculature, NETosis is responsible for increased inflammation/coagulation.
It is also possible that, in turn, inflammation/coagulation activates neutrophils
and induces them to form more NETs either directly or through the consequences of
platelet activation. Would inhibition of coagulation prevent NETosis? Inversely, could
the inhibition of NETs reduce the procoagulant state? Are NETs directly implicated
in metastasis or is the general inflammation and thrombin generation triggered by
NETs the main player?
The discovery of the presence of NETs within the tumor environment also suggests new
leads for the development of diagnostic tools for NET detection and NET inhibition.
For example, PAD4 inhibitors could be used in cancer patients to prevent tumor spread
and cancer associated-thrombosis. Interestingly, the use of PAD inhibitor has already
been assessed in settings of cancer. PAD4 is overexpressed in various human cancers[71]
[72] and it interacts with p53 to repress the transcription of tumor suppressor genes.[73] Therefore, the use of a pan PAD inhibitor[74]
[75] inhibits cancer cell proliferation[73] and reduces tumor growth.[76] Recently, Wang et al have designed a more potent inhibitor of PAD4, which also inhibits
PAD2 to a lesser extent, and observed an even greater inhibition of tumor growth.[77] Thus, in addition to their direct effect on cancer cells, the use of PAD4 inhibitors
would also prevent PAD4-driven NETosis, thereby inhibiting not only tumor growth but
also cancer-associated thrombosis. Of note, as the overexpression of PAD4 in a cancer
cell line leads to the generation of NET-like structures,[17] and PAD4 is markedly overexpressed in many human cancers, the potential release
of cancer cell chromatin, in addition to the neutrophils' extracellular chromatin,
should be considered as it could also affect tumor progression and cancer-associated
thrombosis. The presence of NETs within the tumor microenvironment and in the blood
of tumor-bearing hosts has opened new avenues of research in cancer biology as well
as provided new explanations for the interplay of cancer with inflammation and thrombosis.




