

Subscribe to RSS
DOI: 10.1055/a-2486-6758
Signal Transduction and Transformation by the Platelet Activation Cascade: Systems Biology Insights
Authors
Funding Russian Science Foundation. 23-45-10039

Abstract
Binding of platelet activators to their receptors initiates a signal transduction network, where intracellular signal is filtered, amplified, and transformed. Computational systems biology methods could be a powerful tool to address and analyze dynamics and regulation of the crucial steps in this cascade. Here we review these approaches and show the logic of their use for a relatively simple case of SFLLRN-induced procoagulant activity. Use of a typical model is employed to track signaling events along the main axis, from the binding of the peptide to PAR1 receptor down to the mPTP opening. Temporal dynamics, concentration dependence, formation of calcium oscillations and their deciphering, and role of stochasticity are quantified for all essential signaling molecules and their complexes. The initial step-wise activation stimulus is transformed to a peak at the early stages, then to oscillation calcium spikes, and then back to a peak shape. The model can show how both amplitude and width of the peak encode the information about the activation level, and show the principle of decoding calcium oscillations via integration of the calcium signal by the mitochondria. Use of stochastic algorithms can reveal that the complexes of Gq, in particular the complex of phospholipase C with Gq, which are the limiting steps in the cascade with their numbers not exceeding several molecules per platelet at any given time; it is them that cause stochastic appearance of the signals downstream. Application of reduction techniques to simplify the system is demonstrated.
Keywords
signal transduction - mathematical models - computational systems biology - secondary messengers - reductionIntroduction
Platelet activation is a key event in hemostasis and thrombosis. It transforms a relatively passive resting cell into a novel state capable of performing its major hemostatic functions. The essential physiological platelet activators include adenosine diphosphate (ADP), thrombin, thromboxane A2, and collagen, each binding one or more specific receptors on the plasmatic membrane to initiate signal transduction. The main negative regulators are prostacyclin and NO. Platelet activation is additionally modulated by mechanosensing, contact with fibrinogen and fibrin, epinephrine, and other molecules and conditions. A vast signaling network composed of receptors with their primary effectors, protein kinase cascades, secondary messengers, and signal-deciphering systems ([Fig. 1]) allows the platelet to rapidly process these signals and form physiological responses: shape change, integrin activation, granule release, thromboxane A2 synthesis, and procoagulant activity.[1] [2] The main functions of such networks typically include: (1) filtering out noise and preventing accidental activation; (2) regulation of the activation threshold by positive and negative modulators; (3) transition of the activation signal and its transformation into different degrees of response; (4) regulation of the signal transduction in time (e.g., by inducing refractoriness or stopping transduction after some time); (5) integration of different signals to form a unified response.

Experimental approaches currently can reveal only some of the aspects of this complex network functioning. Numerous unstable complexes formed by intermediate messengers, often membrane-dependent and rapidly changing in time, are tremendously difficult to investigate experimentally. For platelets, this is additionally complicated by their small size and almost complete impossibility to use genetically encoded sensor probes. Computational systems biology methods have been becoming increasingly useful for the investigation of various biological networks, and they can play a particularly important role for analyzing the platelet signal transduction.[3]
Systems Biology of Platelet Signal Transduction: Computational Models and Beyond
Computational modeling has been becoming increasingly popular as a tool for research of complex biochemical systems since the beginning of the 21st century, and the term “computational systems biology” itself was coined around that time. These models allowed in-depth analysis of the biological systems' architecture in relationship with their function.[4] Some theoretical approaches were focused on detailed description of the target systems' biochemistry[5] (as a bottom-up approach), and others used simplified models to capture the essential properties of the same systems, in this case, blood coagulation cascade[6] [7] (top-down approach). Finally, it was possible to combine these approaches for the same system, by systematically going from a detailed model to a simplified one by means of formal reduction employing analysis of sensitivities and temporal hierarchy.[8]
Development of computational systems biology models for platelet signal transduction lagged behind that for blood coagulation following the experimental discoveries. The detailed reviews of the existing models and modeling approaches in the field could be found elsewhere.[3] [9] The pioneering study was that of Purvis et al in 2008,[10] where P2Y1-mediated signal transduction to calcium was simulated. Later computational systems biology studies expanded the list of the studied activators, signaling pathways, and physiological responses.[11] [12] [13] [14] [15] In addition to these detailed mechanism-driven models, neural networks,[16] agent-based approaches,[17] [18] and top-down models of signal transduction[19] were utilized to facilitate simulations, integrate platelet signaling with blood coagulation, and allow use of learning algorithms for individual patients.
Finally, it is important to point out that, in addition to computational simulations, there are other major tools in systems biology, such as causal pathway analysis and others.[20] Recently, they significantly advanced the current understanding of platelet signal transduction. Important examples include transcriptomic-based comparative analysis of signal transduction in mice and humans,[21] and network analysis of genome-wide platelet transcriptome and proteome database.[22]
Although there are important studies and reviews on the subject, easy-to-understand descriptions of the investigation process in computational systems biology of platelet signal transduction are difficult to find. The purpose of this state-of-the-art mini-review is to highlight the details of the process of the analysis of a computational systems biology model for a specific case of platelet functional response. We shall illustrate the utility of this approach to understand every step of platelet signal transduction using a relatively simple example of signaling pathway leading to procoagulant platelet formation upon stimulation by thrombin receptor agonist peptide SFLLRN.[23] The key signaling events along the main axis, from the binding of the peptide to PAR1 receptor down to the mPTP opening, shall be tracked. Temporal dynamics, concentration dependence, formation of calcium oscillations and their deciphering, and role of stochasticity will be quantified at each step. The reduction techniques will be used to simplify the system and explicitly identify key regulators of platelet signal transduction.
Procoagulant Platelets and Signaling
Procoagulant platelets are a subpopulation formed upon sufficiently strong platelet activation and capable of supporting membrane-dependent reactions of blood coagulation on their surface.[24] The phenomenon of the membrane-dependent reactions is ubiquitous in biochemistry, because this makes it possible to achieve high reaction rates with a small number of molecules by concentrating them on a small surface and limiting reaction to two dimensions.[25] There could be other reasons for blood coagulation to adopt the membrane-based approach, such as possibility to protect membrane-bound enzymes from blood flow,[26] or to control fibrin formation by controlling procoagulant platelets' spatial distribution.[27] The systems biology models of blood coagulation network need to consider formation of procoagulant platelets during thrombin generation.[28]
Although some factors such as the resting cytosolic calcium level[13] or the number of mitochondria[13] may predispose a platelet to become procoagulant, the fate of the platelet is ultimately decided in the course of its activation.[29] [30] Depending on the stimulus, between 0 and 100% of resting platelets are capable of becoming procoagulant.[31]
The procoagulant platelet is a terminal state, which is associated with many features of necrosis. It has a typical balloon-shaped appearance,[32] with disrupted membrane integrity and cytoskeleton,[33] and inactivated integrins. Transition to this terminal state is determined by the signal transduction network, where thrombin or collagens are obligatory activators,[34] while weaker ones such as ADP may only modulate the number of procoagulant platelets but not cause them by themselves.[14] Cytosolic calcium and mitochondrial signaling is at the center of procoagulant platelet formation.[35] Platelet activation induces a series of cytosolic calcium spikes, which leads to calcium entry in the mitochondria and opening of mPTP,[36] followed by energy collapse and necrosis. Heterogeneity of platelets and stochastic nature of the platelet signaling results in only part of them becoming procoagulant.[37] The events following permanent mPTP opening are not signaling anymore, because a cell with extremely high cytosolic calcium and no ATP is already dead at this stage. Although these high calcium concentrations activate scramblase TMEM16F and specific calpain isoforms to produce the specific necrotic phenotype, these downstream events are a separate “necrotic biochemistry” story. The mathematical models of signal transduction for procoagulant platelet formation stop at the mPTP opening stage,[37] as the downstream events are already irreversible.
Procoagulant response is a relatively simple example because we only need to consider the main signaling axis from receptor binding to calcium mobilization to mitochondria. Although it may be modulated by other agonists and antagonists, this cascade is completely capable of functioning by itself, and we shall henceforth consider it.
The Model for the Procoagulant Platelet Response
To illustrate the logic of platelet signal transduction, we shall use a mechanism-based computational model of platelet signal transduction originally developed specifically for SFLLRN-induced procoagulant platelet formation.[37] The model includes a number of reactions shown in [Fig. 1], although we shall predominantly analyze the main steps beginning from PAR1 and ending with mPTP opening. When the opening is irreversible, it is equivalent to a platelet becoming procoagulant.[36] Technically, the model is a set of ordinary differential equations (ODEs), where every variable is concentration of a molecule, written in the form:

where Ai is the concentration of a species, and F + and F − are the rates of reactions where it is produced or destroyed respectively, based on the physical and chemical kinetics laws. The model explicitly included several compartments such as cytosol and mitochondria, where changes of the concentrations were considered. Others, such as extracellular space and dense tubular system (DTS), were not analyzed in such detail because we assumed that the concentrations of species there were constant (we did not consider potent activation leading to DTS depletion).
The detailed description of the model design, validation, and principles can be found elsewhere.[3] [11] [12] [14] [37] [38] [39] Briefly, it included 29 species with concentrations described by 27 differential equations, and two species were fixed (see their list and initial values in [Table 1]). All reactions, along with the equations and parameters, are collected in [Table 2]. The model formally included four compartments: cytosol (3 fL), plasmatic membrane equivalent (0.6 pL), DTS (1.5 fL), and mitochondria (0.3 fL). One important aspect to be mentioned is that platelets are small (the total volume of about 5 fL) and therefore the number of certain molecules in the cytosol is sometimes also small. One can easily estimate that 10 nM of calcium ions for a 3 fL large cytosol means 10 × 10−9 × 6 × 1023 × 3 × 10−15 = 18. At many steps of signal transduction, there could be just single molecules. In these cases, ODEs are not applicable anymore, and stochastic algorithms are used to simulate the same chemical system.
|
Species |
Variable (constant) |
Compartment |
Initial condition (µM) |
|---|---|---|---|
|
Cytosolic calcium |
|
Cytosol |
0.013 |
|
IP3 |
|
Cytosol |
0.05 |
|
IP3 state IP3Ra |
|
Cytosol |
0.024 |
|
IP3 state IP3Ri1 |
|
Cytosol |
0.036 |
|
IP3 state IP3Ri2 |
|
Cytosol |
0 |
|
IP3 state IP3Rn |
|
Cytosol |
0.3 |
|
IP3 state IP3Ro |
|
Cytosol |
0.24 |
|
IP3 state IP3Rs |
|
Cytosol |
0 |
|
Mitochondrial membrane potential |
|
Cytosol |
138.7 |
|
Calcium in DTS |
|
DTS |
1000 |
|
Mitochondrial calcium |
|
Mitochondrion |
0.1 |
|
Closed mPTP |
|
Mitochondrion |
1 |
|
Open mPTP |
|
Mitochondrion |
0 |
|
Resting PAR |
|
Plasma membrane |
0.006 |
|
Activated PAR |
|
Plasma membrane |
0 |
|
PAR with a free Gq |
|
Plasma membrane |
0 |
|
PAR with GqGDP |
|
Plasma membrane |
0 |
|
PAR with GqGTP |
|
Plasma membrane |
0 |
|
PIP2 |
|
Plasma membrane |
200 |
|
Phospholipase C (PLC), inactive |
|
Plasma membrane |
0.06 |
|
PLC with GqGDP |
|
Plasma membrane |
0 |
|
PLC with GqGTP |
|
Plasma membrane |
0 |
|
PLCGqGTP with its substrate PIP2 |
|
Plasma membrane |
0 |
|
GDP |
|
Plasma membrane |
0 |
|
Gq with GDP |
|
Plasma membrane |
0.0043 |
|
Gq with GTP |
|
Plasma membrane |
0 |
|
GTP |
|
Plasma membrane |
1 |
|
External thrombin |
|
N/a (extracellular, parameter) |
Indicated in the figures |
|
External calcium |
|
N/a (extracellular, parameter) |
2,000 µM |





























|
Module |
Name |
Reaction |
Flux, compartment |
Parameters |
Ref. |
|---|---|---|---|---|---|
|
PAR |
Activation of PAR1 |
|
|
|
[49] |
|
PAR |
Degradation of PAR1* |
|
|
|
[37] |
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
|
|
|
[49] |
|
|
PAR |
Simplified PI turnover |
|
|
|
[37] |
|
PM |
Calcium PM leak |
|
|
|
[37] |
|
PM |
PMCA |
|
|
|
[37] |
|
DTS |
SERCA2b |
|
|
|
[37] |
|
DTS |
SERCA3 |
|
|
|
[37] |
|
DTS |
Calcium DTS leak |
|
|
|
[37] |
|
DTS |
Calcium release through IP3R |
|
|
|
[49] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
|
[50] |
|
DTS |
IP3R |
|
|
[50] |
|
|
PM |
SOCE |
|
|
|
[51] |
|
DTS |
SOCE |
|
|
|
[51] |
|
PM |
SOCE |
|
|
|
[37] |
|
Mit (*) |
NCLX |
|
|
|
[52] |
|
Mit (*) |
Uniporter |
|
|
|
[53] |
|
Mit |
Uniporter |
|
|
|
[53] |
|
Mit |
NCLX |
|
|
|
[52] |
|
Mit |
Respiratory chain |
|
|
|
[37] |
|
Mit |
mPTP |
|
|
|
[37] |
|
Mit |
mPTP |
|
|
|
[53] |





















































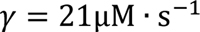





















































It should be clearly stated that this model was chosen for the review purposes because of its simplicity. It does not include a number of later computational systems biology developments, such as feedback phospholipase C activation,[38] numerous mitochondrial compartments, or effects of mPTP opening on platelet energetics;[13] and none of the existing quantitative models include thousands of reactions potentially involved in platelet calcium signaling.[20] All the models should be therefore used specifically for the purpose of hypothesis testing. Any model is a simplification and biological models cannot ever exclude possibility of some undiscovered reactions or molecules. Therefore, the results of computational models should be viewed similarly to the experimental models: as predictions, whose validity should be repeatedly tested.
The signal transformation during platelet activation will be considered in the following order. First, the signal shape transformation will be reviewed. Second, transformation of the signal information will be analyzed. Side-by-side with these steps, a comparison with stochastic calculation will be made. Finally, the possibility of reducing part of the system in order to speed up calculations and reduce the number of model parameters will be performed.
Transformation of the Signal Shape
The transformation of the signal shape during platelet activation by different thrombin concentrations in the deterministic case is shown in [Fig. 2]. The activator concentration at time 0 changes abruptly to a given value, which is essentially a step-type signal shape (or Heaviside step function). We can see decreasing levels of intact-free PAR1 ([Fig. 2A]) and concentration-dependent increase of the active PAR1 ([Fig. 2B]). Afterwards, the signal is transformed into a clear peak-type shape at the PAR1-Gq-GDP complex level ([Fig. 2C]), which is then retained for all intermediate variables including IP3 concentration ([Fig. 2D–K]) up to the calcium ion concentration.

The calcium ion concentration can also acquire a peak-type shape at low stimulation levels, but oscillations are observed at high levels ([Fig. 2L]). However, at the next step, for the mitochondrial calcium concentration, the signal shape again has a peak-type shape ([Fig. 2M]), and mPTP opening follows it ([Fig. 2N]). Thus, it can be expected that there is a system for integrating the oscillatory calcium signal. Indeed, it can be shown that the maximum of the time integral of the calcium ion concentration steadily increases with increasing activator concentration ([Fig. 2O]).
In the case of stochastic integration, individual curves do not carry the same meaning. However, it is possible to consider the averaging of stochastic curves ([Fig. 3]). If we also consider the standard deviation of the variable value, we can evaluate the deviations of the signal shape. As can be seen from the calculation results, the signal transformation preserves the same type of transformation from “step” to a peak “peak.” This holds even in the case when the concentration of a substance is very low and individual runs of the model give only values 0 or 1 molecules. In this case, the average concentration of calcium ions also acquires the form of a “peak,” which is consistent with known experimental data.[40]

This figure is also quite informative with regard to the identification of the “limiting” steps in the signal transduction cascade. One may notice that the curve for PAR1 is smooth ([Fig. 3A]) because there are more than 1,000 PAR1 molecules per platelet. However, the number of active PAR1 at any given time does not exceed 10 molecules per cell ([Fig. 3B]), and all complexes with Gq are present at the level of not more than 4 molecules per cell ([Fig. 3C–I]). The real low point is Gq-bound phospholipase C that has not yet bound substrate: its concentration does not exceed one molecule per cell ([Fig. 3I]). Downstream from it, there are much more molecules but the “damage” has been done by that time: the level of stochasticity beginning from the PLC-Gq-PIP2 complex is huge, with average numbers greatly different from the individual runs ([Fig. 3J–M]).
Transformation of the Signal Information
The peak-shaped signal carries at least two pieces of information in the form of two characteristic parameters, the peak amplitude and the peak width ([Fig. 4]). In order to understand how they “encode” the information about the activator concentration along the signaling cascade, the dependences of these parameters on thrombin concentration were constructed. For all variables of the system, the peak amplitude steadily increased with the activation level, and the peak half-width steadily decreased ([Fig. 4B–N]). Thus, it is possible that the ratio of these variables is a constant value and does not depend on activation. When the value of IP3 of 0.16 μM is exceeded, an oscillatory regime may occur. This value can be considered as a threshold. The longer the IP3 concentration exceeds the specified value, the longer the oscillations will last in the system. From this point of view, the peak half-width is a true characteristic of the signal. On the other hand, the dependence of the maximum calcium concentration in the mitochondrial matrix on the thrombin concentration is linear, while the half-width is not ([Fig. 4M]). However, it should be noted that the peak half-width directly depends on the maximum, while the duration of being above the threshold concentration does not directly depend on it.

To illustrate the question of how the signal is integrated by mitochondria, [Fig. 5] shows a typical stochastic dependence of the calcium concentration in the mitochondrial matrix on the calcium concentration in the cytosol for a model in which a second thrombin receptor was added.

Reduction of the Model
The receptor module of the computational model describes the transmission of a signal from a receptor to the concentration of IP3. This module is relatively isolated, meaning that there is little feedback from the downstream, so it can be analyzed independently. As can be seen from the previous analysis, the shape of the signal does not change along the cascade, nor does the way the signal “encodes” information. This means that a reduction of the corresponding system of equations without loss of information is possible. As a first step, only first-order terms with regard to the resting state were retained in the original model.[39] The equations of the module took the form:

where x 1 is the concentration of the resting PAR1 receptor, x 2 is that of the activated receptor, x 3 is the complex of the activated receptor with the inactivated G protein, x 4 is the activated complex of the receptor with the G protein, x 5 is the intermediate complex of the receptor with the G protein, s 1 is inactive phospholipase C, s 2 is phospholipase C in complex with the inactive G protein, s 3 is activated phospholipase C, s 4 is the complex of activated phospholipase C with its substrate, s 5 is the activated G protein, and s ip3 is the product of phospholipase C (IP3). The variables are reduced for the case of a thrombin concentration of 100 nM.
As can be seen from system (2), the first equation can be solved directly:

where T is the thrombin concentration. The variable s 1 (the concentration of inactivated phospholipase C) changes very slowly and can be equated to 1, while several phospholipase complexes (s 2 to s 4) are, on the contrary, a subsystem of very fast variables converging to a quasi-stationary solution in times of the order of 0.01 s. The exact proof of this can be produced using Tikhonov's theorem and rules for slow–fast dynamics; examples of such approaches are described in detail for other systems.[8] [41] [42] As a result, the equation for s 4, the complex of activated phospholipase C with its substrate, can be written as follows:

where s 4 actually reflects the dynamics of s 5 (GqGTP). While x 2 is a very fast variable, the three variables x 3, x 4, and x 5 form an intermediate group of successive signal transformations with characteristic times of ∼0.5 s. When considering larger characteristic times, they can also be reduced to a single equation for x 2:

Taking into account [eq. 3], we obtain the following solution for x2 :

This solution has the same properties as the original solution ([Fig. 6]). Given the dependence of x 4 on x 2 used here, [eq. 4] for s 4 (this variable now expresses the GqGTP pool) is also solved:


Similarly, the formula for the relative concentration of IP3 is obtained:

The values of parameters A, B, C, and D depend on the concentration of thrombin and the number of receptors (x 10), as well as on the initial concentration of IP3, which is generally zero. Therefore, the following general dependence of the IP3 concentration on time can be assumed:

where x ip3 is the relative concentration of inositol-3-phosphate, Thr is the concentration of the activator (thrombin), [PAR] is the initial concentration of receptors to this activator, A, B, and α are some nonnegative parameters, and f() is some monotonic function. Importantly, such a reduction is not applicable for stochastic integration of the model, since, as a result of it, the variable that determines the stochastic nature of the system's behavior (the amount of the active form of G protein) leaves the model.
The reduction of the calcium signaling block has been repeatedly carried out in various studies.[43] [44] [45] [46] [47] The equations of the last, mitochondrial block are as follows:

The ability of this model to predict the proportion of platelets that pass into a procoagulant state is comparable to that observed in the experiment,[13] [14] [37] [39] [48] indicating that the molecular mechanisms of mitochondrial collapse embedded in the model operate comparably to real living systems. This mechanism consists of the accumulation of calcium ions in the mitochondrial matrix with an increase in the calcium concentration in the cell cytosol ([Fig. 5B]) due to the opening of a uniporter channel. Since the membrane potential changes slowly during the process of calcium accumulation in mitochondria, its values can be considered constant. Then the equation for the calcium concentration in mitochondria is as follows:

where  is the concentration of free calcium ions in the mitochondrial matrix,
is the concentration of free calcium ions in the mitochondrial matrix,  is the concentration of free calcium ions in the cell cytosol, Δψ is the potential difference on the inner mitochondrial membrane, Δψ* is the critical value of the potential difference, and α, γ, K, L are the model parameters. This equation reflects the mechanism of calcium ion accumulation
in the matrix: when the value of K is exceeded, calcium ions enter the matrix at a rate proportional to the parameter
α, while they leave the mitochondria at a rate monotonically dependent on . The maximum concentration of calcium ions in the mitochondrial matrix during this
process is also proportional to the external stimulus. If exceeds K often enough or for a long enough time, the mitochondria become overloaded with calcium
ions and open mPTP permanently.
is the concentration of free calcium ions in the cell cytosol, Δψ is the potential difference on the inner mitochondrial membrane, Δψ* is the critical value of the potential difference, and α, γ, K, L are the model parameters. This equation reflects the mechanism of calcium ion accumulation
in the matrix: when the value of K is exceeded, calcium ions enter the matrix at a rate proportional to the parameter
α, while they leave the mitochondria at a rate monotonically dependent on . The maximum concentration of calcium ions in the mitochondrial matrix during this
process is also proportional to the external stimulus. If exceeds K often enough or for a long enough time, the mitochondria become overloaded with calcium
ions and open mPTP permanently.
Concluding Remarks
In this review, we used a simple systems biology model[37] to illustrate some of the basic principles behind the architecture of the platelet signaling network produced by the computational studies of the last two decades,[3] [10] [13] [14] [15] [18] [37] [38] [39] [48] [49] as well as the tools used for dealing with these systems. A computational model of a biological network could be explored by two major complementary approaches, either direct simulation of the signaling events with every detail or utilization of sensitivity analysis and temporal hierarchy to simplify and reduce the system in order to derive analytical rules.
These approaches allow one to shed some additional light on what is likely to occur once thrombin binds its receptor. One can see initial transformation and encoding of the signal in the form of a peak, whose amplitude and duration encode information about thrombin concentration. Due to the small size of the platelet and to the overall tendency of the cell to minimize the number of signaling proteins when possible, the early stages of the process are extremely stochastic funneling down at the level of phospholipase C and G protein complex. Although the number of molecules per platelet at this level is counted in single digits, encoding information not only in the amplitude, but also in the duration, allows the cell to transmit it reliably. The level of phospholipase C catalyzing the formation of inositol-3-phosphate is essential: the concentration of IP3 relatively accurately reflects the concentration of the activator (except for filtering out the noise, which prevents activation by thrombin below a definite threshold[38]). Then IP3 activates calcium ion channels of the dense tubular system, whose activity nonlinearly depends on both IP3 and the concentration of calcium ions in the cytosol. Thus, the channels and carriers of the DTS membrane can be considered as a next decision point: at a low concentration of IP3 in the platelet cytosol, a slight increase in the calcium concentration is observed, the height of which reflects the initial signal. This might play a role in weaker physiological responses, but is not sufficient to overcome a threshold to enter mitochondria. At a high concentration of IP3 in the platelet cytosol, a series of calcium concentration spikes occurs, with the initial signal being encoded by the number and total duration of spikes. In the case of a series of calcium spikes, there are at least two integrating systems in the system, namely mitochondria for procoagulant activity [37] and calcium-sensitive proteins of the cytosol such as CalDAGGEFI for integrin activation.[12] They function similarly, by capturing calcium ions above certain threshold and retaining them. This converts the oscillating signal back into the peak shape. For the procoagulant platelet formation pathway, with a sufficiently high number of calcium spikes per unit time, the concentration of calcium ions in the mitochondrial matrix increases, which leads to mitochondrial collapse and platelet necrosis. The system of calcium ion accumulation in the mitochondrial matrix is the ultimate decision point in the fate of the cell upon activation.
A number of points remain beyond the scope of this review. The most interesting of them include differences in the signaling principles between the G-protein-dependent pathways[10] [49] and tyrosine kinase-dependent ones,[11] [15] [18] interaction of the signals from different receptors and interactions between pathways mediated by different messengers,[14] [39] simulation of other platelet functional responses such as integrin activation.[12] The current research has just touched upon these problems. A deeper analysis of the physiological meaning of the signal encoding and decoding, regulation of the thresholds and of the response amplitudes for different physiological responses of the platelet, optimization of the signaling system in terms of the cost-efficiency balance, involvement of the membrane-dependent reactions, and spatial aspects of the platelet signal transduction are just some of the exciting subjects, where future prospects for the computational systems biology methods are significant. From the methodological point of view, integration of computational systems biology models with proteomic data[2] [20] [22] is a highly promising line of research and model personalization. Finally, the neural networks approach could be combined with ODE-based models to significantly promote versatility of their use.[16] [19]
Conflict of Interest
A.N.S. and M.A.P. report that the study was supported by the Russian Science Foundation grant 23-45-10039.
Acknowledgments
The authors acknowledge support by the Russian Science Foundation grant 23-45-10039.
-
References
- 1 Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost 2016; 14 (05) 918-930
- 2 Huang J, Zhang P, Solari FA. et al. Molecular proteomics and signalling of human platelets in health and disease. Int J Mol Sci 2021; 22 (18) 22
- 3 Dunster JL, Panteleev MA, Gibbins JM, Sveshnikova AN. Mathematical techniques for understanding platelet regulation and the development of new pharmacological approaches. Methods Mol Biol 2018; 1812: 255-279
- 4 Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 2003; 15 (02) 221-231
- 5 Hockin MF, Jones KC, Everse SJ, Mann KG. A model for the stoichiometric regulation of blood coagulation. J Biol Chem 2002; 277 (21) 18322-18333
- 6 Beltrami E, Jesty J. Mathematical analysis of activation thresholds in enzyme-catalyzed positive feedbacks: application to the feedbacks of blood coagulation. Proc Natl Acad Sci U S A 1995; 92 (19) 8744-8748
- 7 Ataullakhanov FI, Guria GT, Sarbash VI, Volkova RI. Spatiotemporal dynamics of clotting and pattern formation in human blood. Biochim Biophys Acta 1998; 1425 (03) 453-468
- 8 Panteleev MA, Balandina AN, Lipets EN, Ovanesov MV, Ataullakhanov FI. Task-oriented modular decomposition of biological networks: trigger mechanism in blood coagulation. Biophys J 2010; 98 (09) 1751-1761
- 9 Nechipurenko DY, Shibeko AM, Sveshnikova AN, Panteleev MA. In silico hemostasis modeling and prediction. Hamostaseologie 2020; 40 (04) 524-535
- 10 Purvis JE, Chatterjee MS, Brass LF, Diamond SL. A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood 2008; 112 (10) 4069-4079
- 11 Martyanov AA, Balabin FA, Dunster JL, Panteleev MA, Gibbins JM, Sveshnikova AN. Control of platelet CLEC-2-mediated activation by receptor clustering and tyrosine kinase signaling. Biophys J 2020; 118 (11) 2641-2655
- 12 Martyanov AA, Morozova DS, Sorokina MA. et al. Heterogeneity of integrin αIIbβ3 function in pediatric immune thrombocytopenia revealed by continuous flow cytometry analysis. Int J Mol Sci 2020; 21 (09) 21
- 13 Obydennyi SI, Artemenko EO, Sveshnikova AN. et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica 2020; 105 (04) 1095-1106
- 14 Shakhidzhanov SS, Shaturny VI, Panteleev MA, Sveshnikova AN. Modulation and pre-amplification of PAR1 signaling by ADP acting via the P2Y12 receptor during platelet subpopulation formation. Biochim Biophys Acta 2015; 1850 (12) 2518-2529
- 15 Dunster JL, Mazet F, Fry MJ, Gibbins JM, Tindall MJ. Regulation of early steps of GPVI signal transduction by phosphatases: a systems biology approach. PLOS Comput Biol 2015; 11 (11) e1004589
- 16 Chatterjee MS, Purvis JE, Brass LF, Diamond SL. Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli. Nat Biotechnol 2010; 28 (07) 727-732
- 17 Garzon Dasgupta AK, Martyanov AA, Filkova AA, Panteleev MA, Sveshnikova AN. Development of a simple kinetic mathematical model of aggregation of particles or clustering of receptors. Life (Basel) 2020; 10 (06) 10
- 18 Tantiwong C, Dunster JL, Cavill R. et al. An agent-based approach for modelling and simulation of glycoprotein VI receptor diffusion, localisation and dimerisation in platelet lipid rafts. Sci Rep 2023; 13 (01) 3906
- 19 Anand M, Lee M, Diamond S. Combining data-driven neural networks of platelet signalling with large-scale ODE models of coagulation. Sadhana 2018; 43: 1-9
- 20 Babur Ö, Luna A, Korkut A. et al. Causal interactions from proteomic profiles: molecular data meet pathway knowledge. Patterns (N Y) 2021; 2 (06) 100257
- 21 Balkenhol J, Kaltdorf KV, Mammadova-Bach E. et al. Comparison of the central human and mouse platelet signaling cascade by systems biological analysis. BMC Genomics 2020; 21 (01) 897
- 22 Huang J, Swieringa F, Solari FA. et al. Assessment of a complete and classified platelet proteome from genome-wide transcripts of human platelets and megakaryocytes covering platelet functions. Sci Rep 2021; 11 (01) 12358
- 23 London FS, Marcinkiewicz M, Walsh PN. PAR-1-stimulated factor IXa binding to a small platelet subpopulation requires a pronounced and sustained increase of cytoplasmic calcium. Biochemistry 2006; 45 (23) 7289-7298
- 24 Podoplelova NA, Nechipurenko DY, Ignatova AA, Sveshnikova AN, Panteleev MA. Procoagulant platelets: mechanisms of generation and action. Hamostaseologie 2021; 41 (02) 146-153
- 25 Ahmad SS, London FS, Walsh PN. The assembly of the factor X-activating complex on activated human platelets. J Thromb Haemost 2003; 1 (01) 48-59
- 26 Tosenberger A, Ataullakhanov F, Bessonov N, Panteleev M, Tokarev A, Volpert V. Modelling of platelet-fibrin clot formation in flow with a DPD-PDE method. J Math Biol 2016; 72 (03) 649-681
- 27 Nechipurenko DY, Receveur N, Yakimenko AO. et al. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler Thromb Vasc Biol 2019; 39 (01) 37-47
- 28 Susree M, Panteleev MA, Anand M. Coated platelets introduce significant delay in onset of peak thrombin production: Theoretical predictions. J Theor Biol 2018; 453: 108-116
- 29 Veuthey L, Aliotta A, Bertaggia Calderara D, Pereira Portela C, Alberio L. Mechanisms underlying dichotomous procoagulant COAT platelet generation-a conceptual review summarizing current knowledge. Int J Mol Sci 2022; 23 (05) 23
- 30 Alberio L, Ravanat C, Hechler B, Mangin PH, Lanza F, Gachet C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb Haemost 2017; 117 (06) 1101-1114
- 31 Yakimenko AO, Verholomova FY, Kotova YN, Ataullakhanov FI, Panteleev MA. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys J 2012; 102 (10) 2261-2269
- 32 Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 1997; 90 (07) 2615-2625
- 33 Artemenko EO, Yakimenko AO, Pichugin AV, Ataullakhanov FI, Panteleev MA. Calpain-controlled detachment of major glycoproteins from the cytoskeleton regulates adhesive properties of activated phosphatidylserine-positive platelets. Biochem J 2016; 473 (04) 435-448
- 34 Fernández DI, Kuijpers MJE, Heemskerk JWM. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021; 32 (07) 863-871
- 35 Millington-Burgess SL, Harper MT. Cytosolic and mitochondrial Ca2+ signaling in procoagulant platelets. Platelets 2021; 32 (07) 855-862
- 36 Jobe SM, Wilson KM, Leo L. et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008; 111 (03) 1257-1265
- 37 Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol Biosyst 2015; 11 (04) 1052-1060
- 38 Balabin FA, Sveshnikova AN. Computational biology analysis of platelet signaling reveals roles of feedbacks through phospholipase C and inositol 1,4,5-trisphosphate 3-kinase in controlling amplitude and duration of calcium oscillations. Math Biosci 2016; 276: 67-74
- 39 Sveshnikova AN, Balatskiy AV, Demianova AS. et al. Systems biology insights into the meaning of the platelet's dual-receptor thrombin signaling. J Thromb Haemost 2016; 14 (10) 2045-2057
- 40 Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000; 39 (18) 5458-5467
- 41 Shibeko AM, Panteleev MA. Slow-fast dynamics in non-linear enzyme cascades gives rise to spatial multiscaling. Chaos Solitons Fractals 2024; 188: 115594
- 42 Zaitsev AV, Martinov MV, Vitvitsky VM, Ataullakhanov FI. Rat liver folate metabolism can provide an independent functioning of associated metabolic pathways. Sci Rep 2019; 9 (01) 7657
- 43 Li YX, Rinzel J. Equations for InsP3 receptor-mediated [Ca2+]i oscillations derived from a detailed kinetic model: a Hodgkin-Huxley like formalism. J Theor Biol 1994; 166 (04) 461-473
- 44 Magnus G, Keizer J. Model of beta-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am J Physiol 1998; 274 (04) C1158-C1173
- 45 De Young GW, Keizer J. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc Natl Acad Sci U S A 1992; 89 (20) 9895-9899
- 46 Sneyd J, Falcke M. Models of the inositol trisphosphate receptor. Prog Biophys Mol Biol 2005; 89 (03) 207-245
- 47 Sneyd J, Dufour JF. A dynamic model of the type-2 inositol trisphosphate receptor. Proc Natl Acad Sci U S A 2002; 99 (04) 2398-2403
- 48 Obydennyy SI, Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J Thromb Haemost 2016; 14 (09) 1867-1881
- 49 Lenoci L, Duvernay M, Satchell S, DiBenedetto E, Hamm HE. Mathematical model of PAR1-mediated activation of human platelets. Mol Biosyst 2011; 7 (04) 1129-1137
- 50 Pokhilko AV, Ataullakhanov FI, Holmuhamedov EL. Mathematical model of mitochondrial ionic homeostasis: three modes of Ca2+ transport. J Theor Biol 2006; 243 (01) 152-169
- 51 Liu W, Tang F, Chen J. Designing dynamical output feedback controllers for store-operated Ca2+ entry. Math Biosci 2010; 228 (01) 110-118
- 52 Hussain JF, Mahaut-Smith MP. Reversible and irreversible intracellular Ca2+ spiking in single isolated human platelets. J Physiol 1999; 514 (Pt 3): 713-718
- 53 Gillespie DT. Stochastic simulation of chemical kinetics. Annu Rev Phys Chem 2007; 58: 35-55
Address for correspondence
Publication History
Received: 22 October 2024
Accepted: 22 November 2024
Article published online:
19 February 2025
© 2025. Thieme. All rights reserved.
Georg Thieme Verlag KG
Oswald-Hesse-Straße 50, 70469 Stuttgart, Germany
-
References
- 1 Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost 2016; 14 (05) 918-930
- 2 Huang J, Zhang P, Solari FA. et al. Molecular proteomics and signalling of human platelets in health and disease. Int J Mol Sci 2021; 22 (18) 22
- 3 Dunster JL, Panteleev MA, Gibbins JM, Sveshnikova AN. Mathematical techniques for understanding platelet regulation and the development of new pharmacological approaches. Methods Mol Biol 2018; 1812: 255-279
- 4 Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 2003; 15 (02) 221-231
- 5 Hockin MF, Jones KC, Everse SJ, Mann KG. A model for the stoichiometric regulation of blood coagulation. J Biol Chem 2002; 277 (21) 18322-18333
- 6 Beltrami E, Jesty J. Mathematical analysis of activation thresholds in enzyme-catalyzed positive feedbacks: application to the feedbacks of blood coagulation. Proc Natl Acad Sci U S A 1995; 92 (19) 8744-8748
- 7 Ataullakhanov FI, Guria GT, Sarbash VI, Volkova RI. Spatiotemporal dynamics of clotting and pattern formation in human blood. Biochim Biophys Acta 1998; 1425 (03) 453-468
- 8 Panteleev MA, Balandina AN, Lipets EN, Ovanesov MV, Ataullakhanov FI. Task-oriented modular decomposition of biological networks: trigger mechanism in blood coagulation. Biophys J 2010; 98 (09) 1751-1761
- 9 Nechipurenko DY, Shibeko AM, Sveshnikova AN, Panteleev MA. In silico hemostasis modeling and prediction. Hamostaseologie 2020; 40 (04) 524-535
- 10 Purvis JE, Chatterjee MS, Brass LF, Diamond SL. A molecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood 2008; 112 (10) 4069-4079
- 11 Martyanov AA, Balabin FA, Dunster JL, Panteleev MA, Gibbins JM, Sveshnikova AN. Control of platelet CLEC-2-mediated activation by receptor clustering and tyrosine kinase signaling. Biophys J 2020; 118 (11) 2641-2655
- 12 Martyanov AA, Morozova DS, Sorokina MA. et al. Heterogeneity of integrin αIIbβ3 function in pediatric immune thrombocytopenia revealed by continuous flow cytometry analysis. Int J Mol Sci 2020; 21 (09) 21
- 13 Obydennyi SI, Artemenko EO, Sveshnikova AN. et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica 2020; 105 (04) 1095-1106
- 14 Shakhidzhanov SS, Shaturny VI, Panteleev MA, Sveshnikova AN. Modulation and pre-amplification of PAR1 signaling by ADP acting via the P2Y12 receptor during platelet subpopulation formation. Biochim Biophys Acta 2015; 1850 (12) 2518-2529
- 15 Dunster JL, Mazet F, Fry MJ, Gibbins JM, Tindall MJ. Regulation of early steps of GPVI signal transduction by phosphatases: a systems biology approach. PLOS Comput Biol 2015; 11 (11) e1004589
- 16 Chatterjee MS, Purvis JE, Brass LF, Diamond SL. Pairwise agonist scanning predicts cellular signaling responses to combinatorial stimuli. Nat Biotechnol 2010; 28 (07) 727-732
- 17 Garzon Dasgupta AK, Martyanov AA, Filkova AA, Panteleev MA, Sveshnikova AN. Development of a simple kinetic mathematical model of aggregation of particles or clustering of receptors. Life (Basel) 2020; 10 (06) 10
- 18 Tantiwong C, Dunster JL, Cavill R. et al. An agent-based approach for modelling and simulation of glycoprotein VI receptor diffusion, localisation and dimerisation in platelet lipid rafts. Sci Rep 2023; 13 (01) 3906
- 19 Anand M, Lee M, Diamond S. Combining data-driven neural networks of platelet signalling with large-scale ODE models of coagulation. Sadhana 2018; 43: 1-9
- 20 Babur Ö, Luna A, Korkut A. et al. Causal interactions from proteomic profiles: molecular data meet pathway knowledge. Patterns (N Y) 2021; 2 (06) 100257
- 21 Balkenhol J, Kaltdorf KV, Mammadova-Bach E. et al. Comparison of the central human and mouse platelet signaling cascade by systems biological analysis. BMC Genomics 2020; 21 (01) 897
- 22 Huang J, Swieringa F, Solari FA. et al. Assessment of a complete and classified platelet proteome from genome-wide transcripts of human platelets and megakaryocytes covering platelet functions. Sci Rep 2021; 11 (01) 12358
- 23 London FS, Marcinkiewicz M, Walsh PN. PAR-1-stimulated factor IXa binding to a small platelet subpopulation requires a pronounced and sustained increase of cytoplasmic calcium. Biochemistry 2006; 45 (23) 7289-7298
- 24 Podoplelova NA, Nechipurenko DY, Ignatova AA, Sveshnikova AN, Panteleev MA. Procoagulant platelets: mechanisms of generation and action. Hamostaseologie 2021; 41 (02) 146-153
- 25 Ahmad SS, London FS, Walsh PN. The assembly of the factor X-activating complex on activated human platelets. J Thromb Haemost 2003; 1 (01) 48-59
- 26 Tosenberger A, Ataullakhanov F, Bessonov N, Panteleev M, Tokarev A, Volpert V. Modelling of platelet-fibrin clot formation in flow with a DPD-PDE method. J Math Biol 2016; 72 (03) 649-681
- 27 Nechipurenko DY, Receveur N, Yakimenko AO. et al. Clot contraction drives the translocation of procoagulant platelets to thrombus surface. Arterioscler Thromb Vasc Biol 2019; 39 (01) 37-47
- 28 Susree M, Panteleev MA, Anand M. Coated platelets introduce significant delay in onset of peak thrombin production: Theoretical predictions. J Theor Biol 2018; 453: 108-116
- 29 Veuthey L, Aliotta A, Bertaggia Calderara D, Pereira Portela C, Alberio L. Mechanisms underlying dichotomous procoagulant COAT platelet generation-a conceptual review summarizing current knowledge. Int J Mol Sci 2022; 23 (05) 23
- 30 Alberio L, Ravanat C, Hechler B, Mangin PH, Lanza F, Gachet C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb Haemost 2017; 117 (06) 1101-1114
- 31 Yakimenko AO, Verholomova FY, Kotova YN, Ataullakhanov FI, Panteleev MA. Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys J 2012; 102 (10) 2261-2269
- 32 Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 1997; 90 (07) 2615-2625
- 33 Artemenko EO, Yakimenko AO, Pichugin AV, Ataullakhanov FI, Panteleev MA. Calpain-controlled detachment of major glycoproteins from the cytoskeleton regulates adhesive properties of activated phosphatidylserine-positive platelets. Biochem J 2016; 473 (04) 435-448
- 34 Fernández DI, Kuijpers MJE, Heemskerk JWM. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021; 32 (07) 863-871
- 35 Millington-Burgess SL, Harper MT. Cytosolic and mitochondrial Ca2+ signaling in procoagulant platelets. Platelets 2021; 32 (07) 855-862
- 36 Jobe SM, Wilson KM, Leo L. et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008; 111 (03) 1257-1265
- 37 Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol Biosyst 2015; 11 (04) 1052-1060
- 38 Balabin FA, Sveshnikova AN. Computational biology analysis of platelet signaling reveals roles of feedbacks through phospholipase C and inositol 1,4,5-trisphosphate 3-kinase in controlling amplitude and duration of calcium oscillations. Math Biosci 2016; 276: 67-74
- 39 Sveshnikova AN, Balatskiy AV, Demianova AS. et al. Systems biology insights into the meaning of the platelet's dual-receptor thrombin signaling. J Thromb Haemost 2016; 14 (10) 2045-2057
- 40 Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 2000; 39 (18) 5458-5467
- 41 Shibeko AM, Panteleev MA. Slow-fast dynamics in non-linear enzyme cascades gives rise to spatial multiscaling. Chaos Solitons Fractals 2024; 188: 115594
- 42 Zaitsev AV, Martinov MV, Vitvitsky VM, Ataullakhanov FI. Rat liver folate metabolism can provide an independent functioning of associated metabolic pathways. Sci Rep 2019; 9 (01) 7657
- 43 Li YX, Rinzel J. Equations for InsP3 receptor-mediated [Ca2+]i oscillations derived from a detailed kinetic model: a Hodgkin-Huxley like formalism. J Theor Biol 1994; 166 (04) 461-473
- 44 Magnus G, Keizer J. Model of beta-cell mitochondrial calcium handling and electrical activity. I. Cytoplasmic variables. Am J Physiol 1998; 274 (04) C1158-C1173
- 45 De Young GW, Keizer J. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc Natl Acad Sci U S A 1992; 89 (20) 9895-9899
- 46 Sneyd J, Falcke M. Models of the inositol trisphosphate receptor. Prog Biophys Mol Biol 2005; 89 (03) 207-245
- 47 Sneyd J, Dufour JF. A dynamic model of the type-2 inositol trisphosphate receptor. Proc Natl Acad Sci U S A 2002; 99 (04) 2398-2403
- 48 Obydennyy SI, Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J Thromb Haemost 2016; 14 (09) 1867-1881
- 49 Lenoci L, Duvernay M, Satchell S, DiBenedetto E, Hamm HE. Mathematical model of PAR1-mediated activation of human platelets. Mol Biosyst 2011; 7 (04) 1129-1137
- 50 Pokhilko AV, Ataullakhanov FI, Holmuhamedov EL. Mathematical model of mitochondrial ionic homeostasis: three modes of Ca2+ transport. J Theor Biol 2006; 243 (01) 152-169
- 51 Liu W, Tang F, Chen J. Designing dynamical output feedback controllers for store-operated Ca2+ entry. Math Biosci 2010; 228 (01) 110-118
- 52 Hussain JF, Mahaut-Smith MP. Reversible and irreversible intracellular Ca2+ spiking in single isolated human platelets. J Physiol 1999; 514 (Pt 3): 713-718
- 53 Gillespie DT. Stochastic simulation of chemical kinetics. Annu Rev Phys Chem 2007; 58: 35-55