Key words
green synthesis - indoles - aqueous medium - ultrasound - alternative energy
1
Introduction
Green chemistry has provided facile, sustainable protocols with minimal or zero use
of hazardous substances in the manufacturing and application of important chemicals.[1] One of the principles of green chemistry particularly focuses on the use of alternative
energy resources to accomplish chemical reactions. The use of ultrasound waves (US)
as an alternative energy source has grown considerably in various fields.[2] The acoustic cavitation process involved in US promoted methodologies generates
huge amounts of energy due to high local temperature and pressure.[3] The sonochemical reactions proceed via the formation and adiabatic collapse of transitory cavitation bubbles in the liquid
phase.[4a]
[b] A lot of work has been done in this direction and it has been proved that US irradiation
could facilitate the facile construction of biologically relevant organic moieties.[4c–i]
Nature’s solvent, water, has some fascinating structural and physicochemical features
that lead to some particular intermolecular interactions such as hydrogen bonding,
hydrophobic effects, and trans-phase interactions. These interactions lead to several
energy changes during the course of a reaction.[5] However, the effective use of water as reaction medium requires incorporation of
additives in order to homogenize mixtures of water and organic compounds.[6a] The use of US energy resource and aqueous media together can improve the applicability
of both.[6b] The US waves travels through aqueous reaction medium and generates microcavitation
bubbles. The cavitation bubbles are thought to act as micro reactors for the reactants.
The reactant molecules enter into the micro bubbles and the instantaneous high local
temperature and pressure produced during cavitations (due to formation and adiabatic
collapse of cavitation bubbles) not only enhances the chances of reactants coming
closer to each other but also lets the reactants overcome the potential energy barrier,
required for a reaction to occur, very speedily (Figure [1]).[7] In this manner, ultrasound-promoted aqueous-mediated organic synthesis helps in
achieving the goal of green synthesis.
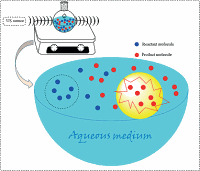 Figure 1 Representative diagram of typical acoustic cavitation process in aqueous medium
Figure 1 Representative diagram of typical acoustic cavitation process in aqueous medium
Most of the known synthetic and naturally occurring drugs and other bioactive compounds
have a key heterocycle structural unit.[8] Furthermore, heterocycles are widely used in various fields such as dyestuffs, fluorescent
materials, luminescent sensors, brightening agents, and analytical reagents.[9] They are also of great significance due to their wide utility as protecting groups,
chiral auxiliaries, organocatalysts and ligands in asymmetric catalysts.[10] Particularly, the indole nucleus is associated with various biological activities
viz. antibacterial, antifungal, antiviral, antitumor, anti-inflammatory, antidepressant,
antimalarial, and anti-HIV agents. A large number of synthetic indoles have profound
applications as pharmaceuticals and agrochemicals.[11] Representatives of bioactive indoles are depicted in Figure [2].
 Figure 2 Representatives of biologically important compounds containing the indole unit
Figure 2 Representatives of biologically important compounds containing the indole unit
Therefore, the synthesis and functionalization of indoles has been an interesting
object for researchers in the current epoch. Recently, extensive efforts have been
undertaken to develop more efficient routes to synthesize biologically vital indole-based
compounds.[12] This review focuses on the combined use of ultrasound and aqueous media for the
facile synthesis of various important indole derivatives.
Synthesis of Biologically Vital Indoles
2
Synthesis of Biologically Vital Indoles
2.1
Spirocyclic Indoles
A library of spiro[indoline-3,4′-pyrazolo[3,4-b]pyridine]-2,6′(1′H)-diones 4a–p was synthesized by Bazgir and co-workers through a three-component reaction of 4-hydroxycoumarin,
1H-pyrazol-5-amines, and isatins in water under US irradiation using p-TSA as catalyst (Scheme [1]).[13] This one-pot protocol involves the formation of two C–C bonds and one C–O bond,
and produces a series of spirooxindole-fused pyrazolopyridine derivatives. The authors
performed the reaction at various temperatures (40, 50, and 60 °C), and found 60 °C
to be a suitable temperature for the conversion of reactants into the desired product
under ultrasonication in water.
 Scheme 1 Synthesis of spiro[indoline-3,4′-pyrazolo[3,4-b]pyridine]-2,6′(1′H)-diones
Scheme 1 Synthesis of spiro[indoline-3,4′-pyrazolo[3,4-b]pyridine]-2,6′(1′H)-diones
Dandia and co-workers have done a lot of work on the synthesis of spirooxindoles and
other indole derivatives. A library of spiro[indole-thiazolidinone] derivatives 8a–uu was synthesized by Dandia et al., via a US-promoted aqueous-mediated route. The approach involved the tandem reaction of
isatins, 2-mercaptopropionic acid, and heterocyclic amines and proceeded in the presence
of cetyltrimethylammonium bromide (CTAB) phase-transfer catalyst. A variety of heterocyclic
cores including dimethyl-2-phenyl-pyrazol-3-one, triazole, benzimidazole, benzothiazole,
and indazole, bearing a primary amino group were utilized as precursors in this protocol
(Scheme [2]).[14] The surfactant CTAB in water enhanced the effect of US waves in accelerating the
reaction, as it acts as an emulsifying agent and forms colloidal dispersion with organic
substrates, consequently, rendering the mixture more homogeneous. Further, the authors
summarized that the use of a high-intensity ultrasonic (HIU) direct immersion probe
as US irradiation source provided better results in terms of yield and time than obtained
with a low-intensity ultrasonic (LIU) cleaner.
 Scheme 2 Synthesis of spiro[indole-thiazolidinone] derivatives
Scheme 2 Synthesis of spiro[indole-thiazolidinone] derivatives
Further, various spiro(indoline-3,4′-pyrano[2,3-c]pyrazole) derivatives 12a–l were synthesized through a three-component US-promoted reaction of substituted isatins,
active methylene reagent, and 3-methyl-1-phenyl-2-pyrazolin-5-one in aqueous medium
using cerium(IV) ammonium nitrate (CAN) as catalyst (Scheme [3]).[15] The acoustic cavitation stimulates the sonolysis and creation of bonds with the
help of the Ce based catalyst.
 Scheme 3 Synthesis of spiro(indoline-3,4′-pyrano[2,3-c]pyrazole) derivatives
Scheme 3 Synthesis of spiro(indoline-3,4′-pyrano[2,3-c]pyrazole) derivatives
Moreover, the authors also checked the antioxidant activities of the as-synthesized
spiroindolinones and the data were compared with standard drug ascorbic acid. They
used synthetic nitrogen-centered DPPH•, ABTS•+ and NO radicals as indicator compounds in the experimental studies. The authors found
that the as-synthesized spiroindoline derivatives showed good radical scavenging abilities;
however, their abilities were moderately lower than those of ascorbic acid. The combination
of carboxylate group with N-benzyl substitution in the indole ring showed excellent
DPPH• radical-scavenging activity.
The authors also proposed a plausible mechanism in this work (Scheme [4]). Accordingly, the acoustic cavitation played an important role in accelerating
the CAN-catalyzed reaction through its induced shear forces and the jets produced
near the surface of the vessel. The reaction involves formation of the isatiylidene
malanonitrile intermediate and its further attack at the C-4 site of 3-methyl-1-phenyl-2-pyrazolin-5-one
followed by intramolecular nucleophilic attack of the -OH group on the cyano moiety.
 Scheme 4 Plausible mechanism for the CAN-catalyzed synthesis of spiro(indoline-3,4′-pyrano[2,3-c]pyrazole) derivatives
Scheme 4 Plausible mechanism for the CAN-catalyzed synthesis of spiro(indoline-3,4′-pyrano[2,3-c]pyrazole) derivatives
Another one-pot, three-component synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] and spiro[chromene-4,3′-indoline] derivatives 16a–l was achieved by Dandia and co-workers using ZnS nanoparticles as catalyst under ultrasonic
irradiation in aqueous medium. Isatins, active methylene reagent, and 3-methyl-1-phenyl-2-pyrazolin-5-one
or dimedone were used as precursor in this protocol (Scheme [5]).[16] The authors employed a variety of isatin substrates bearing electron-withdrawing
and electron-releasing groups, and found that this protocol provided great yields
with both.
 Scheme 5 Synthesis of spiro[chromene-4,3′-indoline] derivatives
Scheme 5 Synthesis of spiro[chromene-4,3′-indoline] derivatives
Dandia and co-workers also explored the vital use of NaCl salt in water under ultrasonication
for the efficient one-pot synthesis of spiro[chromene-4,3′-indolines] 20a–l
via the reaction of isatins, active methylene component (malononitrile or ethylcyanoacetate),
and dimedone (Scheme [6]).[17] All the reactions were performed at ambient temperature. The authors further developed
the utility of NaCl and synthesized various spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives through the same protocol using 3-methyl-1-phenyl-2-pyrazolin-5-one
instead of dimedone. Besides producing a huge amount of energy, the acoustic cavitation
effect here could contribute to the site-specific activation of substrate molecules
by NaCl in water.
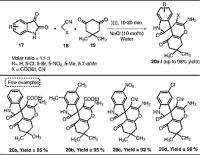 Scheme 6 NaCl catalyzed synthesis of spiro[chromene-4,3′-indolines] derivatives
Scheme 6 NaCl catalyzed synthesis of spiro[chromene-4,3′-indolines] derivatives
A catalyst-free aqueous-mediated multicomponent domino (MCR) protocol was also developed
by Dandia et al. This method involves the chemo- and regio-selective synthesis of spiro[indoline-3,4′-pyrazolo[3,4-e][1,4]thiazepine]diones 25a–i
via three-component reaction of isatin, 5-amino-3-methylpyrazole, and α-mercaptocarboxylic
acid in water under US irradiation (Scheme [7]).[18] The reaction proceeded via Baylis–Hillman type adduct 24 intermediate formation. The authors interestingly explained the efficiency of water
over other solvents used in this protocol under US irradiation by considering the
I
max (maximum cavitation intensity) and T
Imax (the temperature at which I
max is reached) parameters. Further, the authors tested the synthesized compounds for
their α-amylase inhibition activity. Among the spiro[pyrazolo[3,4-e][1,4]thiazepine] derivatives synthesized in this work, compound 25c showed the best results.
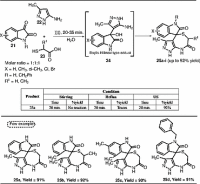 Scheme 7 Synthesis of spiro[indoline-3,4′-pyrazolo[3,4-e][1,4]thiazepine]diones
Scheme 7 Synthesis of spiro[indoline-3,4′-pyrazolo[3,4-e][1,4]thiazepine]diones
In the proposed mechanism, the authors well explained the plausible role of water
in promoting the reaction. Water could create hydrogen bonds with C3-carbonyl functionality
of isatin to a reasonable extent and increase the C3 electrophilicity of the isatin.
Here, water works as a Brønsted acid and results in the rapid formation of Baylis–Hillman
type adduct 24 (Scheme [8]). Further, the water can act as Brønsted base by connecting its oxygen site with
the hydrogen of the thiol group and accelerate the Michael addition reaction of intermediate
A with the thioacid, followed by dehydration.
 Scheme 8 Plausible mechanism for the catalyst-free synthesis of spiro[indoline-3,4′-pyrazolo[3,4-e][1,4]thiazepine]diones
Scheme 8 Plausible mechanism for the catalyst-free synthesis of spiro[indoline-3,4′-pyrazolo[3,4-e][1,4]thiazepine]diones
Arya et al. reported the synthesis of spiro[indole-pyrido[3,2-e]thiazine] derivatives 29a–j
via the reaction of various isatins with amine and 2-mercaptonicotinic acid catalyzed
by ZSM-5-([MIM]+BF4
−) Brønsted acid ionic liquid catalyst system in water under US irradiation (Scheme
[9]). The authors proved the effectiveness of US in aqueous medium over conventional
and microwave irradiation methods. Further, they explained the particular role of
water as reaction medium under ultrasonication in this reaction by giving the example
of the formation of highly reactive radicals that are responsible for the rapid organic
transformation.[19]
 Scheme 9 Synthesis of spiro[indole-pyrido[3,2-e]thiazine] derivatives
Scheme 9 Synthesis of spiro[indole-pyrido[3,2-e]thiazine] derivatives
As proposed by the authors (Scheme [10]), the generation of imine diradical A
in situ by the reaction of substituted isatin 26 and aromatic amines 27 while being subjected to sonication is the key step in the mechanism. Further, diradical
intermediate A reacts with 2-mercaptonicotinic acid 28 to give the desired product via diradical intermediate B.
 Scheme 10 Plausible mechanism for the synthesis of spiro[indole-pyrido[3,2-e]thiazine] derivatives
Scheme 10 Plausible mechanism for the synthesis of spiro[indole-pyrido[3,2-e]thiazine] derivatives
Various spiro[4H-pyrano[3,2-c]quinoline-4,3′-indoline]-2′,5(6H)-dione derivatives 33a–j were prepared by Holizadeh and Radmoghadam. They performed the reaction of 4-hydroxy-2H-quinolin-2-one, malononitrile or ethyl cyanoacetate and isatins in water assisted
by US irradiation using piperidine as catalyst (Scheme [11]).[20] The authors also carried out the reaction without sonication in water and they observed
that the reaction proceeded in 80 minutes while the use of US irradiation decreased
the reaction time by 16 times. Probably the application of US increased the solvability
of water to provide the better results. The authors also observed that the amidic
N-H group of 4-hydroxy-2H-quinolone did not have any significant effect on the course of reaction, since N-substituted
and unsubstituted quinolones participated similarly in the reaction.
 Scheme 11 Synthesis of spiro[4H-pyrano[3,2-c]quinoline-4,3′-indoline]-2′,5(6H)-dione derivatives
Scheme 11 Synthesis of spiro[4H-pyrano[3,2-c]quinoline-4,3′-indoline]-2′,5(6H)-dione derivatives
According to the proposed mechanism, the reaction proceeds with the formation of intermediate
isatylidenemalononitrile A by the condensation of isatin 30 and malononitrile 31. Then, 4-hydroxyquinolin-2(1H)-one 32 moiety attacks intermediate A in a Michael type addition manner, at its exocyclic C=C bond; this is then followed
by an intramolecular cyclization to give desired product 33 (Scheme [12]).
 Scheme 12 Plausible mechanism for the synthesis of spiro[4H-pyrano[3,2-c]quinoline-4,3′-indoline]-2′,5(6H)-diones
Scheme 12 Plausible mechanism for the synthesis of spiro[4H-pyrano[3,2-c]quinoline-4,3′-indoline]-2′,5(6H)-diones
The efficacy of US waves in organic synthesis has also been demonstrated by Naeimi
et al. by providing a one-pot synthetic protocol for various tetrahydrospiro[chromene-4,3′-indoline]
derivatives 37a–h using sulfonated chitosan-coated Fe3O4 nanoparticles (Fe3O4@CS-SO3H NPs) in aqueous media (Scheme [13]). Various isatins, malononitrile, and 1,3-dicarbonyl compounds were used as precursors
in this reaction.[7b] The authors performed the experiment at a range of US frequencies and found 35 KHz
to be optimal in terms of reaction yield and time parameters.
 Scheme 13 Synthesis of tetrahydrospiro[chromene-4,3′-indoline] derivatives
Scheme 13 Synthesis of tetrahydrospiro[chromene-4,3′-indoline] derivatives
Hojati and co-workers developed a US-assisted, catalyst-free, room-temperature protocol
for the synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives 42a–l
via the four-component reaction of ethyl acetoacetate, hydrazine hydrate or phenylhydrazine,
isatins, and malononitrile in aqueous media (Scheme [14]).[21] They proved the effective role of US in a noncatalyzed route over a catalyzed pathway.
The reaction proceeded smoothly in just 5 minutes with isatin but required some more
time in the case of substituted isatins.
 Scheme 14 Synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
Scheme 14 Synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
A protocol involving the Knoevenagel–Michael cyclization of isatins, malononitrile
and α-isotriocyanatoimide to afford various 3,3′-pyrrolidonyl-spirooxindoles 46a–p in water under US irradiation at room temperature has been reported by Miao et al. (Scheme [15]).[22] They explained that sonication is responsible for rapid dispersion of solids on
the surface of water and, consequently, enhancement of contact between molecules.
 Scheme 15 Synthesis of 3,3′-pyrrolidonyl-spirooxindole derivatives
Scheme 15 Synthesis of 3,3′-pyrrolidonyl-spirooxindole derivatives
The authors also proposed a mechanism (Scheme [16]) for the above protocol that involves the formation of dicyanoalkene intermediate
A by Knoevenagel condensation of isatin 43 with malononitrile 44. In the next step, Michael addition of the Knoevenagel adduct A with the enolate intermediate B, formed from α-isothiocyanatoimide 45, to form intermediate C. This intermediate C undergoes an intramolecular cyclization to afford the desired product 46 via intermediate D.
 Scheme 16 Plausible mechanism for the synthesis of 3,3′-pyrrolidonyl-spirooxindoles
Scheme 16 Plausible mechanism for the synthesis of 3,3′-pyrrolidonyl-spirooxindoles
Liju et al. reported a one-pot, four-component reaction of substituted phenylhydrazine, dialkyl
acetylenedicarboxylate, substituted isatins, and malononitrile in water/ethanol medium
catalyzed by l-proline (Scheme [17]). This ultrasound-promoted, room-temperature protocol provides an extensive range
of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives 51a–r. Interestingly, the authors noted a change in the temperature of both the water bath
and the reaction system during the reaction under ultrasonication.[23]
 Scheme 17 Synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
Scheme 17 Synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
As proposed by authors (Scheme [18]), intermediate A, which is formed through the initial reaction of 47 and 48, undergoes enolization in the presence of l-proline to give B. Additionally, intermediate C is generated by the Knoevenagel condensation between 49 and 50. Further, l-proline catalyses the deprotonation of intermediate B and its Michael type addition to C to give intermediate D. This intermediate D produces the desired product 51 through intramolecular cyclization.
 Scheme 18 Plausible mechanism for the l-proline catalyzed synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
Scheme 18 Plausible mechanism for the l-proline catalyzed synthesis of spiro[indoline-3,4′-pyrano[2,3-c]pyrazole] derivatives
 Scheme 19 Synthesis of spiropyrano[2,3-c]pyrazole carboxylate derivatives and spiro[indoline-3,4′-pyridine] derivatives
Scheme 19 Synthesis of spiropyrano[2,3-c]pyrazole carboxylate derivatives and spiro[indoline-3,4′-pyridine] derivatives
A series of spiro-oxindoles has been synthesized by Ghomi and co-workers in water
using US as an energy resource at room temperature (Scheme [19]).[24] They performed one-pot multicomponent reaction of various isatins, dimethylacetylenedicarboxylate
(DMAD), malononitrile, and various hydrazine hydrate or anilines in order to prepare
spiro-oxindoles including spiropyrano[2,3-c]pyrazole carboxylate derivatives 56a–l and spiro[indoline-3,4′-pyridine] derivatives 58a–l. They used a supported molybdenum complex on cross-linked poly(1-aminopropyl-3-vinylimidazolium
bromide) entrapped cobalt oxide nanoparticles (Co3O4@PPIL-Mo) as catalyst. In this report, the authors demonstrated and explained the
synergistic effect of US waves, water, and active catalytic sites. US irradiation
facilitated electron-transfer in the reaction environment of aqueous media. Further,
the collapse of cavitation bubbles provided localized hotspots that helped the reaction
to occur rapidly.[24]
Non-spiro 3-Substituted Indoles
2.2
Non-spiro 3-Substituted Indoles
A variety of bis(indolyl)methanes 61a–l were synthesized by Li et al. via electrophilic substitution reactions of indoles with aromatic carbonyl compounds
in aqueous medium under ultrasound irradiation (Scheme [20]). This reaction was catalyzed by aminosulfonic acid.[25] Since the reaction involves attack of an electrophile on the indole ring, the reaction
provided better results with aromatic aldehydes bearing electron-withdrawing substituents,
as expected.
 Scheme 20 Synthesis of aryl- and cycloalkyl-substituted bis(indolyl)methanes
Scheme 20 Synthesis of aryl- and cycloalkyl-substituted bis(indolyl)methanes
As per the proposed reaction mechanism (Scheme [21]), the protic acid activates the carbonyl functionality and the produced electrophile
causes an electrophilic substitution reaction at the C3 carbon of indole 59. The formed intermediate A undergoes dehydration to give intermediate B. Further, intermediate B is activated by a proton and serves as an electrophile to attack a second molecule
of indole to form the desired product 61.
 Scheme 21 Plausible mechanism for the synthesis of bis(indolyl)methanes
Scheme 21 Plausible mechanism for the synthesis of bis(indolyl)methanes
The same research group also reported another improved methodology for synthesizing
bis(indolyl)methanes 64a–m. Here, they have used dodecylbenzenesulfonic acid (ABS) as an effective catalyst
for the greener synthesis. In this work they also demonstrated the combined effect
of US irradiation and water for the facile synthesis (Scheme [22]).[26]
 Scheme 22 Synthesis of bis(N-indolyl)methanes
Scheme 22 Synthesis of bis(N-indolyl)methanes
Gill and co-workers prepared a variety of bis(indol-3-yl)methanes 67a–n by the one-pot reaction of indole and various aldehydes in water using ultrasound
irradiation at ambient temperature. They used 1-hexenesulphonic acid sodium salt as
catalyst (Scheme [23]).[27] The authors showed that 1-hexenesulphonic acid sodium salt liberates the corresponding
acid when dissolved in water during the course of the reaction and catalyzes the reaction
to afford the desired product under US irradiation.
 Scheme 23 Synthesis of aryl- and heteroaryl-substituted bis(indolyl)methanes
Scheme 23 Synthesis of aryl- and heteroaryl-substituted bis(indolyl)methanes
Khorshidi and Tabatabaeian have reported the ultrasound promoted synthesis of 3-indolyl-3-hydroxyoxindoles
70a–k in aqueous media by performing the reaction of isatins and indoles catalyzed by Fe(III)
homogeneous catalyst. They reported that all products were solely monoindolylated
isatins not 3,3-di-3-indolylindolin-2-ones (Scheme [24]).[28] The US waves helps in generating fine emulsions and enhances mass transfer in aqueous
medium. The authors also checked the effect of US power (taking the frequency constant)
on reaction commencement and observed that an increase in US power from 20 to 100%
(from 92 to 460 W cm–2, respectively) resulted in a decrease in reaction time. At high power, the cavitation
process is maximized and reactants become distributed throughout the reaction mixture
effectively, which results in completion of reaction in shorter time.
 Scheme 24 Synthesis of 3-indolyl-3-hydroxyoxindole derivatives
Scheme 24 Synthesis of 3-indolyl-3-hydroxyoxindole derivatives
Further, Li and co-workers demonstrated the combined effect of US and aqueous media
for the one-pot Mannich reaction of secondary amine, formaldehyde, and indole or N-methylindole
giving Mannich bases 74a–i related to gramine (Scheme [25]).[29] Further, the authors examined the role of US frequency on this reaction protocol
and found better results in case of 25 kHz US frequency source than with a 40 kHz
frequency source. The lower frequency irradiation can produce better cavitation than
higher frequency.
 Scheme 25 Synthesis of indole-based Mannich bases
Scheme 25 Synthesis of indole-based Mannich bases
The Amrollahi research group reported the synthesis of bis(indole) derivatives 77a–l by the reaction of indole with various electron-deficient alkenes in aqueous media
under ultrasonication using 12-tungstophosphoric acid (H3PW12O40) as catalyst (Scheme [26]).
 Scheme 26 Synthesis of various bis(indole) derivatives
Scheme 26 Synthesis of various bis(indole) derivatives
 Scheme 27 Synthesis of mono-indolyl derivatives
Scheme 27 Synthesis of mono-indolyl derivatives
The expected bis(indole) derivatives were obtained with various 2-benzylidenemalononitrile
and 3-(phenyl)acrylates,[30] but when 2-(pyridylmethylene)malononitriles and 3-(pyridyl)acrylates were used as
electron-deficient alkenes, only the corresponding indolyl derivatives 80a–f were formed (Scheme [27]). The authors presented a reason for this result. Possibly, in the presence of H3PW12O40 acid catalyst, the oxidative dehydrogenation of 2-((indolyl)(pyridyl)-methylene)malononitriles
and of 3-(indolyl)(pyridyl)acrylates catalyst is less reactive than 2-((aryl)(indolyl)methyl)malonitriles
and 3-(aryl)(indolyl)acrylates, respectively, which stops the reaction up to indolyls.
Further, in another work, the authors demonstrated a one-pot, four-component procedure
for condensing indole, aldehydes, and active methylene compounds in aqueous media
to give bis(indole) derivatives 84a–l under ultrasonication using 12-tungstophosphoric acid as catalyst (Scheme [28]).[31]
 Scheme 28 Synthesis of bis(indole) derivatives
Scheme 28 Synthesis of bis(indole) derivatives
Here also, bis(indole) derivatives were obtained in case of various benzaldehydes
but only mono-substituted products 88a–l were obtained with various pyridinecarboxaldehydes (Scheme [29]).
 Scheme 29 Synthesis of mono-indolyl derivatives
Scheme 29 Synthesis of mono-indolyl derivatives
Kaur et al. explored the utility of US and water for the efficient synthesis of a variety of
3-amino alkylated indoles 92a–y (Scheme [30]). They performed the reaction of aromatic aldehydes and aniline derivatives in aqueous
media using US as an alternative energy resource and PLA (polylactic acid) grafted
ZnO nanoparticles as catalyst.[32]
 Scheme 30 Synthesis of 3-amino alkylated indoles
Scheme 30 Synthesis of 3-amino alkylated indoles
Yonemitsu condensation for the preparation of 3-indole derivatives 96a–q was performed by Lu and co-workers (Scheme [31]). They explored the use of US irradiation for facile synthesis in aqueous glycerol
using indoles and Meldrum’s acid as precursor at room temperature without catalyst.[33] The authors reported that aliphatic aldehydes showed lower reactivity in this reaction
than aromatic aldehydes. Particularly for aromatic aldehydes, the ortho-substituted aromatic aldehydes gave lower yields, probably due to the steric hindrance
at the ortho position.
 Scheme 31 Synthesis of 3-substituted indoles
Scheme 31 Synthesis of 3-substituted indoles
Nikpassand et al. utilized various pyrazolecarbaldehydes and developed a protocol for their reaction
with indole to give bis(indolyl)methanes 99a–j under US irradiation using water as reaction medium (Scheme [32]).[34] Pyrazole carbaldehydes bearing electron-withdrawing groups showed higher reactivity
than their electron-releasing counterparts.
 Scheme 32 Synthesis of bis(indolyl)methanes
Scheme 32 Synthesis of bis(indolyl)methanes
Aqueous mediated synthesis of bis(indolyl)methanes 102a–n was reported by Kasar and Thopate using US as an alternative energy resource catalyzed
by malic acid organocatalyst (Scheme [33]). This ambient-temperature protocol involved the reaction of indole and various
aromatic aldehydes.[35]
 Scheme 33 Synthesis of bis(indolyl)methanes using organocatalyst
Scheme 33 Synthesis of bis(indolyl)methanes using organocatalyst
A series of bis(indolyl)methane derivatives 105a–o were prepared by the Deshmukh research group using pyruvic acid catalyst in water
under ultrasonication (Scheme [34]).[36] The authors synthesized 15 compounds by using this protocol and they compared all
the US-promoted reactions with the conventional method. They found that while synthesizing
all these 15 bis(indolyl)methane products, the US-promoted methodology had advantages
over conventional approach in terms of both reaction time and yields.
 Scheme 34 Synthesis of bis(2-alkylindolyl)methanes
Scheme 34 Synthesis of bis(2-alkylindolyl)methanes
The authors also proposed a reaction mechanism involving the pyruvic acid as protic
acid catalyst (Scheme [35]). Essentially, the carbonyl functionality is activated by the protic acid, which
facilitates the electrophilic attack of protonated carbonyl A at C3 carbon of indole 103 and further generation of condensed intermediate B. The desired product 105 is formed by the attack of intermediate B on a second molecule of indole.
 Scheme 35 Plausible mechanism for the pyruvic acid catalyzed synthesis of bis(2-alkylindolyl)methanes
Scheme 35 Plausible mechanism for the pyruvic acid catalyzed synthesis of bis(2-alkylindolyl)methanes
2.3
Miscellaneous Indole Syntheses
Various spiro[indole-oxathiolanes] 108a–f were prepared by Dandia and co-workers in aqueous media under US by the reaction
of spiro[indole-3,2′-oxiranes] with thioacetamide using LiBr as catalyst (Scheme [36]).[37] Here, US energy proved to be a better source than the microwave source. The authors
also explained the role of US in heterogeneous reaction conditions. They emphasized
that both mechanical and chemical effects of sonochemical cavitation are responsible
for the acceleration of reaction rate.
 Scheme 36 Synthesis of spiro[indole-3,2′-oxirane] derivatives
Scheme 36 Synthesis of spiro[indole-3,2′-oxirane] derivatives
The same research group also reported the diastereoselective synthesis of fluorine-containing
spiro[indole-3,2′-oxirane]-3′-benzoyl-2(1H)-ones 110a–g by the epoxidation of 3-aroylmethylene indole-2-one in aqueous medium using US as
energy resource (Scheme [37]).[38]
 Scheme 37 Synthesis of spiro[indole-3,2′-oxirane]-3′-benzoyl-2(1H)-ones
Scheme 37 Synthesis of spiro[indole-3,2′-oxirane]-3′-benzoyl-2(1H)-ones
The authors also screened the synthesized compounds for antimicrobial and antioxidant
activities. They showed that Gram negative bacteria are more susceptible to the as-synthesized
compounds than Gram positive organisms. In general, all the synthesized compounds
showed moderate to good activity against all the bacteria. Particularly, compound
110d showed excellent activity against E. coli. The synthesized spiro[indole-3,2′-oxiranes] derivatives bearing halogen substituents
also showed greater antioxidant properties with respect to NO and DPPH methods.[38]
Ramana and co-workers reported the synthesis of dihydropyrano[2,3-e]indole derivatives 114a–h
via the three-component reaction of aldehydes, malononitrile, and 1-tosyl-1H-indol-4-ol under ultrasonication in water/ethanol solvent system. Barium titanate
nanoparticles (BaTiO3 NPs) were used as catalyst to catalyze this reaction (Scheme [38]).[39] The reaction proceeded more efficiently using US horn than that in a sonication
bath.
 Scheme 38 Synthesis of dihydropyrano[2,3-e]indole derivatives
Scheme 38 Synthesis of dihydropyrano[2,3-e]indole derivatives
As proposed by the authors in the plausible mechanism (Scheme [39]), the metallic part (M
n+) of the metal oxides acts as a Lewis acid whereas oxide (O2–) behaves as a base. The reaction involves initial Knoevenagel condensation of aldehyde
and malononitrile, both activated by BaTiO3 NPs, resulting in the formation of intermediate A. This intermediate A undergoes Michael-type addition with 1-tosyl-1H-indol-4-ol to give intermediate B. Then, intermediate B undergoes Thorpe–Ziegler type cyclization, promoted by both the Lewis acidic and
basic sites of BaTiO3 NPs, followed by tautomerization to give the desired product 114.
 Scheme 39 Plausible mechanism for the synthesis of dihydropyrano[2,3-e]indole derivatives
Scheme 39 Plausible mechanism for the synthesis of dihydropyrano[2,3-e]indole derivatives
A variety of hydrazine carboxamides 117a–l were synthesized by Ahsan et al. in water-glycerol system under US irradiation (Scheme [40]). Isatin and various N-(substituted phenyl)hydrazine carboxamides were used as starting
materials in this protocol.[40] The best results were obtained when water-glycerol (3:2) was used as a reaction
medium under ultrasonication. Further, the authors investigated the synthesized compounds
for in vitro anticancer activity against nine separate panels of 60 cancer cell lines according
to NCI US protocols. On the basis of structure–activity relationship studies, it was
concluded that the compound with 4-chloro substitution on the phenyl ring showed excellent
anticancer activity.[40]
 Scheme 40 Synthesis of N-(substituted phenyl)hydrazine carboxamide derivatives
Scheme 40 Synthesis of N-(substituted phenyl)hydrazine carboxamide derivatives
3
Conclusions
The acoustic cavitation in ultrasound promoted reactions generates a large amount
of local heat and pressure that can assist in rapid conversion of reactants into products.
This accelerated conversion can lead to highly selective synthesis in terms of regio-,
chemo-, and stereoselectivity. Water is one of the safest solvents used in green reaction
processes. The beneficial effects of these two, alternative US energy and Nature’s
solvent water, are enhanced when used together. This combined strategy has been widely
explored for the facile synthesis of biologically important indole derivatives. In
this review article, we have collected the recently reported synthetic methodologies
related to synthesis of indoles, including spiro-, non-spiro-, and other indole-based
molecules, in aqueous medium under ultrasonication. Although, there is rapidly developing
interest in this direction, some points remain to be addressed: (1) There is a large
scope for work in the direction of enantio-selective synthesis of bis(indolyl)-derivatives
in aqueous medium under ultrasonication. (2) New paths may be made possible for the
utilization of two-dimensional carbon materials as catalytic supports by graphene
nanosheets due to their huge specific surface area and presence of reactive oxygen
functional groups. These materials can also be explored for the synthesis of various
spiro- and non-spiro indole derivatives under US in water. (3) A lot of work can also
be done on analyzing the bioactivities of newer indole derivatives synthesized under
US in water.
We sincerely hope that this review article will provide comprehensive information
on the synthesis of indole derivatives in aqueous medium under ultrasonication, and
open new doors for the further development of related processes.




