Results and Discussion
The elaborate molecular design of 1a⊂M ([Scheme 1]) was devised to comprise all the components required for supramolecular switching
in an electrostatic field and its optical observation. The donor–π–acceptor chromophore
serves as both a hydrophobic station for the cyclophane and as a fluorescent readout
for the macrocycleʼs position on the rod. The oligo(ethyleneglycol) (OEG) chain on
the rod ensures sufficient space for the cyclophaneʼs translation along the rod axis,
while the OEG chains on the stoppers are terminated by masked thiols to anchor the
molecule on the gold surface. Both OEG chains further serve to separate the chromophore
from the surface to avoid quenching of the chromophoreʼs excited states.[22],[23] Stoppers at the ends of the rod mechanically interlock the dicationic macrocycle
M. We chose the Diederich-type cyclophane M
[24],[25] due to its permanently charged piperidinium groups, the high association constants
it exhibits in water, its ability to change emission properties of guests, as well
as the ease of analysis by NMR due to its high symmetry and significant changes in
chemical shifts upon complexation.[26]–[30] As additional water-solubilizing groups, anionic sulfonates were considered, pulling
the rod towards the cathode and thereby supporting an upright arrangement of the rotaxane
immobilized on the anode.
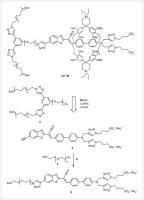 Scheme 1 Design of 1a⊂M and assembly strategy thereof from stopper 2 and chromophore 3. Synthesis of 5 via CuAAC of 3 with 4. Reaction conditions: a) TBTA, Cu(CH3CN)4PF6, sodium ascorbate, DMF/H2O (1 : 1), r. t., o. n., 35%.
Scheme 1 Design of 1a⊂M and assembly strategy thereof from stopper 2 and chromophore 3. Synthesis of 5 via CuAAC of 3 with 4. Reaction conditions: a) TBTA, Cu(CH3CN)4PF6, sodium ascorbate, DMF/H2O (1 : 1), r. t., o. n., 35%.
To take advantage of the antennaʼs near-field enhancement, it is essential for the
chromophore to match the antenna resonance, i.e. emitting light in the red spectral
region. Simultaneously, the chromophore needs to be sufficiently narrow to fit into
the cyclophaneʼs cavity. Accordingly, chromophore 3 was designed to have both a strongly electron-withdrawing group and an electron-donating
group bridged by a biphenyl system. Knoevenagel condensation allowed us to combine
a terminal alkyne-functionalized benzothiazole and a versatile aldehyde precursor
previously synthesized in our group (Scheme S3).[26] The synthesis is described in the Supporting Information. The electron-donating
amine on 3 is decorated with two triazol-propylsulfonate groups, which can act as a mechanical
stopper and a water-solubilizing group simultaneously. As a result, the chromophore
is water soluble and thus suited for rotaxane assembly driven by the hydrophobic effect.[24] This compact design yielded emission maxima above 700 nm in aqueous solution (Figure
S8), paving the way for incorporation in plasmonic nano-antennas.
We decided to test the applicability of this chromophore for the purpose of Au(111)
surface immobilization and molecule–antenna coupling in the gap of the optical antenna
setup. To this end, we attached a thioacetyl-terminated OEG chain (for the synthesis,
see Scheme S2) in a copper(I)-catalyzed azide-alkyne 1,3-cycloaddition (CuAAC) “click”
reaction to give the extended chromophore 5 ([Scheme 1]). The OEG chain was envisioned as spacer to the surface and chosen due to its more
hydrophilic character compared to typically employed alkyl chains in self-assembled
monolayers (SAMs). This allows us to better anticipate potential difficulties in the
rotaxane assembly, which will contain OEG chains as linkers as well. The additional
1,2,3-triazol has a small impact on the optical properties, shifting absorption bathochromically
and emission hypsochromically, but the emission in the red spectral range is preserved
(Figure S8). To deprotect the masked thiol anchoring group and immobilize the molecule
on Au(111), 5 is dissolved in methanol and a solution of sodium methoxide (5.4 M in methanol) is
added. The gold sample is then immersed in this solution for 20 min, washed with water
and dried in a stream of nitrogen gas. We could not observe SAM formation by photoluminescence
(PL) when 5 was immobilized without prior deprotection.[31]
The PL measurements of 5 irradiated on the plasmonic nano-antenna demonstrated that the resulting emission
is enhanced, polarized, and spectrally shaped by the presence of the optical antenna
([Figures 2] and S2).[17],[32]–[34] After chemisorption of 5 on an exemplary antenna ([Figure 2]a), a hyperspectral PL map is recorded by scanning the region of interest with the
excitation laser ([Figure 2]b). Hardly any emission is detected on glass confirming the surface-selective immobilization
of 5 on the Au(111) surface. Furthermore, the local density of states is increased in
the vicinity of the optical antenna (i.e., mostly inside the gap and towards the antenna
ends), resulting in an increased rate of radiative decay of the molecules compared
to the non-radiative decay. Hence, a significantly enhanced emission is observed for
molecules on the antenna when compared to the flat Au(111) surface under saturation
conditions. The high rate of molecule excitation in the antenna gap causes significant
photobleaching within seconds (t
1/2 = 4.1 s) of irradiation with 50 µW 532 nm light under a stream of nitrogen gas (Figure
S1). Nonetheless, the chromophore is sufficiently stable to observe its emission after
30 s of irradiation and is therefore suited for the intended switching experiments.
 Figure 2 Optical characterization of the molecule–antenna system. a) SEM image of an optical antenna with a gap of 30 nm. The plasmonic nano-antenna consists
of the two horizontal gold nanorods in the center of the image. The two vertical wires
are responsible for providing the electrical contact. b) Hyperspectral PL map of the same region as in (a) excited by light of 532 nm wavelength.
The light-emitting SAM of 5 covers only the gold surface and its emission is significantly enhanced by the antenna.
c) PL measurements of nano-antennas of different lengths coated with 5. The PL of the molecule–antenna system (solid lines) is influenced by the scattering
spectrum of the gold nano-antennas (dashed lines), showing the molecule–antenna coupling
(see Supporting Information). The emission maximum of 5 on flat gold is shown as a red-dotted line at 620 nm.
Figure 2 Optical characterization of the molecule–antenna system. a) SEM image of an optical antenna with a gap of 30 nm. The plasmonic nano-antenna consists
of the two horizontal gold nanorods in the center of the image. The two vertical wires
are responsible for providing the electrical contact. b) Hyperspectral PL map of the same region as in (a) excited by light of 532 nm wavelength.
The light-emitting SAM of 5 covers only the gold surface and its emission is significantly enhanced by the antenna.
c) PL measurements of nano-antennas of different lengths coated with 5. The PL of the molecule–antenna system (solid lines) is influenced by the scattering
spectrum of the gold nano-antennas (dashed lines), showing the molecule–antenna coupling
(see Supporting Information). The emission maximum of 5 on flat gold is shown as a red-dotted line at 620 nm.
Due to the shape of the antenna, the enhancement is polarization-dependent and most
photons are polarized along the long axis of the antenna (see Supporting Information).
The resulting emission peak is closer to the strong resonance of the antenna at 650 nm
than the emission polarized along the short axis.
We have repeated these experiments with different antenna lengths to observe the impact
of a change in resonance energy ([Figure 2]c). As expected, the emission enhancement mainly affects transitions in the chromophore
close to the resonance energies of the antenna. The maximum of PL emission is shifted
bathochromically when the antenna scattering maximum is shifted to lower energies.
Moreover, the PL intensity decreases as the resonances of the chromophore and antenna
are further detuned.
These experiments not only show the molecule–antenna coupling and associated emission
enhancement, polarization, and frequency shift, but also that the chromophore fulfils
the stability requirement of the experiment. This axle design thus seems to be ideal
for our intent to immobilize rotaxanes on the antenna and observe the translational
motion of the macrocycle along the molecular axis optically.
Encouraged by these results, we developed the stopper 2 based on previous designs[29],[35] and incorporated an OEG chain with an azide handle for mild rotaxane stoppering
via aqueous CuAAC. Furthermore, in analogy to 5, it comprises masked thiol anchoring groups for its immobilization on a gold surface.
The synthesis of 2 is described in the Supporting Information (Scheme S6) and depicted in [Scheme 4].
The assembly of mechanically interlocked superstructures like 1a⊂M based on the precursor 3 turned out to be challenging. While 3 is soluble in water, mixing it with M·2Cl leads to immediate precipitation of both components out of D2O already at concentrations below 0.5 mM. After unsuccessful screening of the complexation
conditions and a variety of ineffective derivatization attempts to increase the water
solubility of 3, the assembly strategy was revised fundamentally ([Scheme 2]a). The new approach is based on the biphenyl derivative 6, with a twofold functionality acting as a terminal stopper and as a guest for the
cyclophane host M. The plan was to first assemble the pseudorotaxane 6⊂M, which can subsequently be stoppered with 3 in a CuAAC to form 1b⊂M. The design of 6 thus comprises a biphenyl hydrophobic station and a doubly substituted amine as stopper,
similar to 3. The synthesis of 6 is described in the Supporting Information (Scheme S4). Despite the OEG chains, the
extended stopper 6 was not readily soluble in water. However, to our delight, when heated to 60 °C in
the presence of a 2 mM solution of M·2Cl in D2O, the pseudorotaxane 6⊂M was observed by 1H-NMR spectroscopy after cooling back to room temperature (Scheme S1). Encapsulation
of aromatic guests by M·2Cl in D2O is typically associated with strong upfield shifts of the aromatic 1H signals, induced by the aromatic ring current of the cyclophaneʼs walls.[25],[36] The observed shielding of the biphenyl protons (up to 2 ppm) confirms the encircling
by M and the formation of the pseudorotaxane 6⊂M.
 Scheme 2 New rotaxane design inverting the previously employed strategy: A) 3 acts as a stopper, while 6 serves as a rod for pseudorotaxane formation with M·2Cl. Subsequent CuAAC is envisioned to result in mechanically interlocked molecule
1b⊂M. B) Alternative biphenyl 6b combining a rod and a stopper.
Scheme 2 New rotaxane design inverting the previously employed strategy: A) 3 acts as a stopper, while 6 serves as a rod for pseudorotaxane formation with M·2Cl. Subsequent CuAAC is envisioned to result in mechanically interlocked molecule
1b⊂M. B) Alternative biphenyl 6b combining a rod and a stopper.
We set out to synthesize 1b⊂M following this new strategy. First, the pseudorotaxane 6⊂M was formed by heating 6 in water in the presence of excess M·2Cl. Next, CuSO4 and sodium ascorbate were added, followed by chromophore 3 to stopper the pseudorotaxane via CuAAC. While HRMS (Figure S5) and impure 1H-NMR spectra suggested the formation of 1b⊂M as an interlocked species, purification attempts revealed that the [2]rotaxane was
not a stable mechanically interlocked species in organic solvents like DMSO at room
temperature (Figures S6 and S7). Instead an equilibrium between the [2]rotaxane 1b⊂M and its components 1b and M was observed, resulting in the steady degradation of the isolated [2]rotaxane 1b⊂M. Intriguingly, we had previously confirmed that the bulk of the triazol-propylsulfonate
groups on the chromophore suffice to mechanically interlock a rotaxane.[26] We anticipated 6 to be of similar size but probably underestimated the role of the ionic subunits
in the mechanical fixation in solution. However, it appears that the macrocycle can
pass the stopper subunit of 6 already at room temperature. We are currently investigating simplified model compounds
to rationalize and quantify this observation.
The structure comprising the station for the cyclophane was thus redesigned with a
stopper with sufficient steric bulk inspired by 2. Biphenyl 6b was supposed to act as a hydrophobic guest for the cyclophane with a pair of attached
triazoles exposing OEG chains with terminal anchor groups as stoppers. Its synthesis
is presented in the Supporting Information (Scheme S5). In spite of the OEG chains,
the compound 6b turned out to be too insoluble in water to enable the pseudorotaxane formation. Its
assembly and properties thus rather display the challenge to find the subtle balance
between water solubility and self-assembling features driven by the hydrophobic effect.
Our previously synthesized [2]rotaxane based on chromophore 7 showed increased quantum yields upon encapsulation in both water and DMSO, making
it another potential candidate for our endeavor.[26] As a result of the less electron-withdrawing substituent, its absorption and emission
are hypsochromically shifted compared to derivatives of 3. Its optical properties are thus closer to the gold interband absorption, which might
increase the challenges faced in the intended experiment. However, with emission signals
clearly above 550 nm, it should still be observable individually, and compared to
3, 7 showed increased water solubility and was shown to have a high association constant
with M·2Cl in water.[26]
Foregoing the need for a stopper acting as a hydrophobic station, we decided to use
our original stopper 2 for our target [2]rotaxane 8⊂M ([Scheme 3]). The mechanically interlocked molecule was assembled from M·2Cl, 2, and 7 using classical CuAAC “click” reaction conditions with CuSO4 and sodium ascorbate. All reactants were soluble in water, which allowed us to take
full advantage of the hydrophobic effect – the main driving force for the host–guest
complexation with M.[24] Interestingly, within 2 h at room temperature, the solution had lost nearly all
its orange color, and an orange precipitate had formed. This already suggested a significantly
decreased water solubility of 8⊂M and 8 compared to the individual reactants. Purification of the precipitate by reversed-phase
HPLC on C-18 functionalized silica enabled the separation of [2]rotaxane 8⊂M from unthreaded rod 8, and after further purification of 8, the two products were isolated in 21% and 28% yield, respectively. 8⊂M and 8 were fully characterized by 1H-, 13C-, and 2D-NMR spectroscopy, along with HRMS. Both the [2]rotaxane and rod showed
low solubility in water, but good solubility in DMSO and mixtures of ACN and water.
Unlike 1b⊂M, 8⊂M was found to be a room-temperature-stable mechanically interlocked superstructure,
showing neither dissociation in DMSO over time nor during purification by HPLC.
 Scheme 3 Synthesis of 8 and 8⊂M via the CuAAC “click” reaction of stopper 2 and chromophore 7 in aqueous solution. Reaction conditions: a) CuSO4 · 5 H2O, sodium ascorbate, r. t., 2 h, 21% (8⊂M), 28% (8). Comparison of an excerpt of the 1H-NMR spectra of 8 (top, 500 MHz) and 8⊂M (bottom, 600 MHz) in DMSO-d6. Colored dots indicate 1H assignments in 8 and dotted lines show the shifts of these signals in 8⊂M.
Scheme 3 Synthesis of 8 and 8⊂M via the CuAAC “click” reaction of stopper 2 and chromophore 7 in aqueous solution. Reaction conditions: a) CuSO4 · 5 H2O, sodium ascorbate, r. t., 2 h, 21% (8⊂M), 28% (8). Comparison of an excerpt of the 1H-NMR spectra of 8 (top, 500 MHz) and 8⊂M (bottom, 600 MHz) in DMSO-d6. Colored dots indicate 1H assignments in 8 and dotted lines show the shifts of these signals in 8⊂M.
The mechanically interlocked nature of 8⊂M is made apparent by NOE signals between the rod and the cyclophane (see Supporting
Information). Intriguingly, NOEs between the alkyl protons of the triazolpropylsulfonate
groups of the stopper and the protons in proximity to the piperidinium of M are observed (Figure S11). These signals potentially suggest the attraction of the
two oppositely charged water-solubilizing groups. This phenomenon could explain the
stability of 8⊂M compared to 1b⊂M, where one of the stoppers lacks the anionic moiety. The implication of this attraction
on the shuttling behavior in an electrostatic field will be interesting to investigate.
Comparison of the 1H-NMR spectrum of 8⊂M to that of 8 reveals distinct shielding of the encapsulated protons in the isolated 8⊂M ([Scheme 3]). The central protons of the biphenyl system are shifted most significantly compared
to 8 with Δδ of 1.8 and 2.3 ppm, indicating their role as the preferred position of the cyclophane.
Interestingly, the olefinic and neighboring aromatic protons are shifted more (0.5
and 1.2 ppm, respectively) than the outer biphenyl proton next to the amine (~0.3 ppm).
We interpret this as the macrocycle being prevalent on the more electron-deficient
part of the chromophore due to its electron-rich aromatic walls. The 1H signal of the amide is only weakly influenced by the cyclophane and the signals
belonging to either stopper show negligible changes in chemical shift.
For our intended purpose of electrostatic field-driven switching, we require a significant
difference in optical properties of the two states. Thus, the optical properties of
the chromophore in the free rod 8 were compared with the ones of the complexed [2]rotaxane 8⊂M. The absorption maximum of 8⊂M is only slightly bathochromically shifted compared to 8, with 422 versus 415 nm in H2O (Figure S9), and 426 versus 417 nm in DMSO, respectively ([Figure 3]). On the other hand, the emission is shifted hypsochromically in 8⊂M, with maxima at 654 and 635 nm in water and DMSO, respectively, compared to 671 and
652 nm for 8. While the position of the absorption and emission maxima only slightly varied by
encapsulating the chromophore in the rotaxane, a significant difference in the emission
intensity was observed. Quantification was performed by absolute fluorescence quantum
yield measurement in an integrating sphere, corrected for indirect excitation (see
Supporting Information). While the encapsulated chromophore in 8⊂M emitted with an efficiency of 6.5% in water (65.7% in DMSO), the bare chromophore
in 8 showed a quantum yield of only 2.2% in water (20.5% in DMSO). We have previously
observed similar fluorescence enhancements with mechanically interlocked structures
bearing the same chromophore core.[26] These changes in intensity are crucial to observe the electrostatic field-induced
switching in an optical antenna gap.
 Figure 3 Absorption (solid line) and emission (dashed line) spectra of 8⊂M (black) and 8 (red) in DMSO solution. The inset table lists the fluorescence quantum yields of
both molecules in DMSO and H2O.
Figure 3 Absorption (solid line) and emission (dashed line) spectra of 8⊂M (black) and 8 (red) in DMSO solution. The inset table lists the fluorescence quantum yields of
both molecules in DMSO and H2O.
We are delighted to report 8⊂M as a [2]rotaxane fulfilling the boundary conditions for the intended E-field-driven and optically monitored translational switching experiment. Currently
we are optimizing the immobilization conditions on Au(111) surfaces with both 8⊂M and 8.
Experimental Section
Experimental procedures and analytical data for building blocks are provided in the
Supporting Information. Spectra and NMR assignments are given in the Supporting Information.
Macrocycle M was synthesized according to the literature-known procedure.[25] Chromophore 7 was previously described by our group.[26] Fabrication of the gold nano-antenna is briefly described in the Supporting Information.
Reagents and solvents were used as received from commercial suppliers (Sigma-Aldrich,
Acros, Apollo Scientific, Alfa Aesar, Combi-Blocks, Fluorochem, and TCI Chemicals).
Reactions were performed under an argon atmosphere. For column chromatography, Siliaflash
P60 (40 – 63 µm) from Silicycle was used with technical-grade solvents (Biosolve).
TLC was performed on silica gel 60 F254 aluminium sheets from Merck. 1H NMR, 13C{1H} NMR, and 2D NMR spectra were recorded at 298 K on Bruker Avance III NMR spectrometers
equipped with either BBFO, BBI, or BBO probeheads operating at 400 or 500 MHz proton
frequencies. For the rotaxane 8⊂M, NMR spectra were recorded at 298 K on a Bruker Avance III HD NMR spectrometer operating
at 600 MHz proton frequency equipped with a QCI-F probehead. The chemical shifts are
reported in ppm and referenced to the residual solvent peaks, with coupling constants
(J) given in Hz. High-resolution electron spray ionization mass spectrometry (HR-ESI-MS)
spectra were recorded on a maXis 4 G instrument from Bruker. UV-Vis absorption spectra
were recorded on a Jasco V-770 spectrophotometer equipped with a Peltier-thermostatted
cell holder (ETCR-762) set to 25 °C. Emission spectra in solution were recorded on
a Jasco FP-8600 spectrofluorometer equipped with a Peltier-thermostatted cell holder
(ETC-815) set to 25 °C. Quantum yields were determined on the same spectrofluorometer
equipped with a nitrogen-flushed integrating sphere (ILFC-847S). Each measurement
for quantum yield determination (blank solvent, direct excitation, indirect excitation)
was repeated three times and the average of the three calculated quantum yields was
taken.
Synthesis of 5: 3 (97.2 mg, 119 µmol, 1.0 equiv.) and 4 (74.9 mg, 119 µmol, 1.0 equiv.) were dissolved in a mixture of DMF/H2O (1 : 1, 4 mL) and the resulting solution was degassed with argon for 15 min. TBTA
(3.3 mg, 6 µmol, 5 mol%), Cu(CH3CN)4PF6 (2.2 mg, 6 µmol, 5 mol%), and sodium ascorbate (1.2 mg, 6 µmol, 5 mol%) were added
and the reaction mixture was stirred under argon overnight. The reaction mixture was
directly subjected to reverse-phase column chromatography (C18-functionalized silica,
7 : 3 H2O/ACN buffered with 20 mM NH4OAc and 0.05% AcOH). The obtained red-orange solid contained acetamide, which was
removed by two cycles of sonication in EtOH (20 mL) and subsequent centrifugation.
The pure product 5 was obtained as an orange-red solid (60 mg, 35%) after drying in high vacuum.
1H NMR (500 MHz, DMSO-d
6): δ = 8.68 – 8.65 (m, 2 H), 8.39 (s, 1 H), 8.16 – 8.12 (m, 3 H), 8.06 (dd, J = 8.5, 1.7 Hz, 1 H), 8.03 (s, 2 H), 7.87 (d, J = 8.6 Hz, 2 H), 7.68 (d, J = 8.6 Hz, 2 H), 7.05 – 6.92 (m, 8 H) 4.72 (s, 4 H), 4.61 (t, J = 5.1 Hz, 2 H), 4.43 (t, J = 7.1 Hz, 4 H), 3.89 (t, J = 5.2 Hz, 2 H), 3.58 – 3.55 (m, 2 H), 3.53 – 3.43 (m, 46 H), 3.00 (t, J = 6.5 Hz, 2 H), 2.39 (dd, J = 8.2, 6.6 Hz, 4 H), 2.32 (s, 3 H), 2.11 – 2.04 (m, 4 H).
13C NMR (126 MHz, DMSO-d
6): δ = 195.1, 163.8, 152.5, 148.3, 147.7, 145.6, 144.1, 144.0, 135.2, 131.1, 129.7, 128.9,
127.6, 125.9, 125.7, 124.6, 123.4, 123.1, 122.4, 118.5, 116.5, 113.0, 103.3, 69.8,
69.8, 69.7, 69.7, 69.7, 69.6, 69.6, 69.5, 68.9, 68.6, 68.6, 49.8, 48.5, 48.1, 45.6,
37.9, 30.5, 28.3, 26.5.
HRMS-ESI: m/z [M-2NH4 + 5H]3+ calcd for (C62H90 N14O18S4): 471.8378; found: 471.8378; m/z [M-2NH4 + 4H]2+ calcd for (C62H90 N14O18S4): 707.2527; found: 707.2541; m/z [2 M-4NH4 + 7H]3+ calcd for (C62H90 N14O18S4): 942.6679; found: 942.6689; m/z [M-2NH4 + 3H]+ calcd for (C62H90 N14O18S4): 1413.4982; found: 1413.4986.
UV/Vis (H2O): λmax = 271, 445 nm.
Fluorescence (H2O): λmax = 729 nm; Φfl = 0.6% (H2O), 16.7% (DMSO).
Synthesis of 3,5-bis((triisopropylsilyl)ethynyl)phenol (S24): Reaction conditions were adapted from the literature.[37] 3,5-Dibromophenol (CAS Reg. No. 626 – 41 – 5, 1.26 g, 5.00 mmol, 1.0 equiv.), copper
iodide (19 mg, 0.10 mmol, 2 mol%), Pd(PhCN)2Cl2 (77 mg, 0.20 mmol, 4 mol%), and HPtBu3BF4 (117 mg, 0.40 mmol, 8 mol%) were dissolved in anhydrous dioxane (15 mL) under an
argon atmosphere and stirred at r. t. for 30 min. TIPS-acetylene (2.8 mL, 12.50 mmol,
2.5 equiv.) and (iPr)2NH (3.5 mL, 25.0 mmol, 5.0 equiv.) in anhydrous dioxane (10 mL) were added to the
reaction mixture and the resulting mixture was stirred at r. t. overnight. A solution
of HCl (2 M) was added, the product extracted with EtOAc (3×) and the combined organic
layers dried with Na2SO4, filtered, and the solvent evaporated. The crude product was purified by flash chromatography
(gradient cyclohexane to cyclohexane/DCM 70 : 30). The pure product S24 was obtained as a colorless solid (1.96 g, 86%).
1H NMR (400 MHz, CDCl3): δ = 7.15 (t, J = 1.4 Hz, 1 H), 6.90 (d, J = 1.3 Hz, 2 H), 4.78 (s, 1 H), 1.12 (s, 47 H).
13C NMR (101 MHz, CDCl3): δ = 155.0, 128.3, 125.0, 119.2, 105.8, 91.6, 18.8, 11.4.
HRMS-ESI: m/z [M + H]+ calcd for (C28H44OSi2): 453.3014; found: 453.3024.
Synthesis of 3,5-diethynylphenol (S25): S24 (1.96 g, 4.31 mmol, 1.0 equiv.) in an argon-flushed flask was dissolved in TBAF solution
(1 M in THF, 17.2 mL, 17.2 mmol, 4.0 equiv.) under argon and stirred overnight at
room temperature. The solvent was evaporated, the residue was dissolved in TBME and
sat. NH4Cl solution, extracted with TBME (3×) and the extracts washed with sat. NH4Cl solution, water, and brine. The organic layer was dried with Na2SO4, filtered, and the solvent evaporated. Purification by flash chromatography (gradient
cyclohexane to DCM) and subsequent trituration of the solid with n-pentane gave the pure product S25 (406 mg, 66%) as a colorless fluffy solid after filtration and drying in high vacuum.
The analytical data match the previously reported results.[38]
1H NMR (400 MHz, CDCl3): δ = 7.21 (t, J = 1.4 Hz, 1 H), 6.95 (d, J = 1.4 Hz, 2 H), 4.91 (s, 1 H), 3.07 (s, 2 H).
13C NMR (101 MHz, CDCl3): δ = 155.2, 128.7, 123.8, 119.7, 82.4, 78.1.
HRMS-ESI: m/z [M – H]− calcd for (C10H6O): 141.0346; found: 141.0348.
Synthesis of S26: S25 (131.9 mg, 0.928 mmol, 1.0 equiv.) and S13 (540.9 mg, 1.86 mmol, 2.0 equiv.) were dissolved in H2O/acetone (1 : 4, 2.5 mL). CuSO4 · 5 H2O (23.2 mg, 0.093 mmol, 0.1 equiv.) and sodium ascorbate (37.5 mg, 0.186 mmol, 0.2
equiv.) were added under argon. The reaction mixture was stirred at r. t. for 3.5 h.
The mixture was extracted with EtOAc (3×), the extracts washed with brine, dried with
Na2SO4, filtered, and the solvent evaporated. The crude product was purified by flash chromatography
(cyclohexane/acetone 1 : 1) to give the product S26 as a clear pale-yellow oil (575 mg, 86%).
1H NMR (400 MHz, CDCl3): δ = 8.07 (s, 2 H), 7.86 (t, J = 1.4 Hz, 1 H), 7.72 (s, 1 H), 7.45 (d, J = 1.5 Hz, 2 H), 4.59 (t, J = 4.9 Hz, 4 H), 3.94 – 3.87 (m, 4 H), 3.67 – 3.56 (m, 20 H), 2.71 (dd, J = 7.7, 7.0 Hz, 4 H), 1.28 (s, 18 H).
13C NMR (101 MHz, CDCl3): δ = 157.6, 147.5, 132.7, 121.8, 114.9, 112.7, 71.1, 70.7, 70.7, 70.6, 70.3, 69.6,
50.5, 42.3, 31.1, 28.0.
HRMS-ESI: m/z [M + H]+ calcd for (C34H56 N6O7S2): 725.3725; found: 725.3722; m/z [M + Na]+ calcd for (C34H56 N6O7S2): 747.3544; found: 747.3546.
Synthesis of S27: To S26 (105 mg, 145 µmol, 1.0 equiv.), S12 (54 mg, 145 µmol, 1.0 equiv.), and K2CO3 (40 mg, 290 µmol, 2.0 equiv.) in a round-bottom flask was added DMF (2 mL) under
an argon atmosphere. The reaction mixture was heated to 80 °C for 3 d, then allowed
to cool to room temperature and extracted with EtOAc (3×). The extracts were dried
with Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified
by flash chromatography (gradient cyclohexane to cyclohexane/acetone 55 : 45) to give
the pure product S27 as a clear pale-yellow oil (95 mg, 71%).
1H NMR (400 MHz, CDCl3): δ = 8.04 (s, 2 H), 7.87 (t, J = 1.4 Hz, 1 H), 7.42 (d, J = 1.4 Hz, 2 H), 4.60 (t, J = 5.0 Hz, 4 H), 4.27 (t, J = 4.8 Hz, 2 H), 3.96 – 3.87 (m, 6 H), 3.77 – 3.53 (m, 30 H), 3.37 (t, J = 5.1 Hz, 2 H), 2.69 (t, J = 7.3 Hz, 4 H), 1.28 (s, 18 H).
13C NMR (101 MHz, CDCl3): δ = 159.7, 147.4, 132.7, 121.5, 115.7, 111.6, 71.2, 71.0, 70.9, 70.8, 70.8, 70.8,
70.7, 70.7, 70.4, 70.2, 69.9, 69.7, 67.8, 50.8, 50.6, 42.2, 31.1, 28.0.
HRMS-ESI: m/z [M + H]+ calcd for (C42H71 N9O10S2): 926.4838; found: 926.4831; m/z [M + Na]+ calcd for (C42H71 N9O10S2): 948.4658; found: 948.4653.
Synthesis of stopper 2: To a solution of S27 (83 mg, 89.6 µmol, 1.0 equiv.) in anhydrous ACN (1 mL) under argon was added acetyl
chloride (0.68 mL, 9.4 mmol, 105 equiv.), followed by Bi(OTf)3 (60 mg, 89.6 µmol, 1.0 equiv.). The reaction was stirred at r. t. for 5 min, then
water was added, and the mixture was extracted with EtOAc (3 µ). The extracts were
dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified
by reverse-phase HPLC (C-18, ACN/H2O 65 : 35 + 0.1% HCO2H) to give the product 2 as a clear colorless oil (40 mg, 50%). A side product was separated by HPLC and its
analytical data and presumed structure are given in the Supporting Information.
 Scheme 4 Synthesis of stopper 2. Reaction conditions: a) (i-Pr)2NH, CuI,(PhCN)2PdCl2, (t-Bu)3PHBF4, dioxane, r. t., o. n., 86%; b) TBAF, THF, r. t., o. n., 66%; c) CuSO4 · 5 H2O, sodium ascorbate, acetone/H2O (4 : 1), r. t., 3.5 h, 86%; d) K2CO3, DMF, 80 °C, 5 d, 71%; e) AcCl, Bi(OTf)3, ACN, r. t., 5 min, 50%.
Scheme 4 Synthesis of stopper 2. Reaction conditions: a) (i-Pr)2NH, CuI,(PhCN)2PdCl2, (t-Bu)3PHBF4, dioxane, r. t., o. n., 86%; b) TBAF, THF, r. t., o. n., 66%; c) CuSO4 · 5 H2O, sodium ascorbate, acetone/H2O (4 : 1), r. t., 3.5 h, 86%; d) K2CO3, DMF, 80 °C, 5 d, 71%; e) AcCl, Bi(OTf)3, ACN, r. t., 5 min, 50%.
1H NMR (400 MHz, CDCl3): δ = 8.04 (s, 2 H), 7.85 (t, J = 1.5 Hz, 1 H), 7.40 (d, J = 1.4 Hz, 2 H), 4.58 (t, J = 5.0 Hz, 4 H), 4.28 – 4.21 (m, 2 H), 3.93 – 3.86 (m, 6 H), 3.74 – 3.49 (m, 30 H),
3.35 (t, J = 5.1 Hz, 2 H), 3.01 (t, J = 6.5 Hz, 4 H), 2.28 (s, 6 H).
13C NMR (101 MHz, CDCl3): δ = 195.6, 159.7, 147.2, 132.5, 121.5, 115.7, 111.5, 70.9, 70.8, 70.7, 70.7, 70.7,
70.6, 70.6, 70.3, 70.1, 69.8, 69.7, 69.5, 67.7, 50.7, 50.5, 30.6, 28.8.
HRMS-ESI: m/z [M + H]+ calcd for (C38H59 N9O12S2): 838.3797; found: 898.3790; m/z [M + Na]+ calcd for (C38H59 N9O12S2): 920.3617; found: 920.3620; m/z [M + K]+ calcd for (C38H59 N9O12S2): 936.3356; found: 936.3352.
Synthesis of 8⊂M and 8: 7
[26] (8.0 mg, 10.8 µmol, 1.0 equiv.), M·2 Cl[25] (12.7 mg, 11.9 µmol, 1.1 equiv.), and 2 (12.0 mg, 13.4 µmol, 1.2 equiv.) were placed in a 25 mL round-bottom flask and flushed
with argon. Degassed H2O (10 mL) was added, the mixture was stirred for 5 min and CuSO4 · 5 H2O (4.5 mg, 18.0 µmol, 1.7 equiv.) and sodium ascorbate (10.0 mg, 49.5 µmol, 4.6 equiv.)
were added. The mixture immediately turned turbid and showed formation of an orange
precipitate. After stirring at room temperature for 2 h, the precipitate was collected
by centrifugation. The crude product was purified by reverse-phase HPLC on C-18 functionalized
silica (ACN/H2O 40 : 60 + 0.5% HCO2H), allowing the isolation of the pure rotaxane 8⊂M as an orange-red solid (6.0 mg, 21%). The product proved to be poorly soluble in
H2O or ACN, but well soluble in H2O/ACN mixtures. The rod 8 eluted with the macrocycle M and further purification was necessary. Separation on Sephadex LH-20 (DMF/H2O 1 : 1, 100 mM NH4OAc) was unsuccessful. Cyclophane M was removed by trituration of the solid with saturated NH4OAc solution in ACN. The solids were triturated with EtOH to remove excess NH4OA c. The pure side product 8 was obtained as an orange-red solid (4.9 mg, 28%) after drying in high vacuum.
Analytical data for 8⊂M:
1H NMR (600 MHz, DMSO-d6): δ = 8.88 (t, J = 5.7 Hz, 1 H), 8.63 (s, 2 H), 8.06 (s, 2 H), 7.98 (s, 1 H), 7.96 (t, J = 1.5 Hz, 1 H), 7.69 (s, 1 H), 7.38 (d, J = 1.4 Hz, 2 H), 6.85 – 6.74 (m, 10 H), 6.67 (d, J = 8.4 Hz, 2 H), 5.86 (d, J = 8.1 Hz, 2 H), 5.51 (d, J = 7.2 Hz, 2 H), 4.67 (s, 4 H), 4.58 (t, J = 5.2 Hz, 4 H), 4.54 – 4.48 (m, 8 H), 4.22 (t, J = 4.5 Hz, 2 H), 3.88 (t, J = 5.2 Hz, 4 H), 3.83 (t, J = 5.3 Hz, 2 H), 3.80 (t, J = 4.6 Hz, 2 H), 3.72 – 3.19 (m, 92 H, expected: 40 H, overlaps with H2O, H4), 3.04 – 2.97 (m, 8 H), 2.94 (t, J = 6.5 Hz, 4 H), 2.44 (t, J = 7.2 Hz, 4 H), 2.29 (s, 6 H), 2.16 – 2.10 (m, 4 H), 1.25 – 1.12 (m, 24 H).
13C NMR (151 MHz, DMSO-d6): δ = 195.1, 161.1, 159.4, 152.9, 150.7, 147.8, 145.9, 144.4,
144.3, 143.4, 134.9, 132.8, 130.1, 127.5, 126.8, 125.3, 124.5, 123.5, 123.2, 122.3,
116.9, 114.4, 112.3, 110.4, 103.1, 101.9, 71.0, 70.0, 69.8, 69.7, 69.7, 69.6, 69.6,
69.5, 69.0, 68.8, 68.8, 68.6, 67.3, 55.6, 55.2, 55.0, 49.7, 49.3, 48.5, 47.9, 45.7,
42.9, 35.4, 30.5, 28.2, 27.7, 26.6, 25.2, 7.0.
HRMS-ESI: m/z [M + 3H]3+ calcd for (C127H174 N20O31S4): 868.7252; found: 868.7268; m/z [M + Na]+ calcd for (C127H174 N20O31S4): 1302.5841; found: 1302.5861.
UV/Vis: λmax = 422 nm (H2O), 426 nm (DMSO).
Fluorescence: λmax = 654 nm (H2O), 635 nm (DMSO); Φfl = 6.5% (H2O), 65.7% (DMSO).
Analytical data for 8:
1H NMR (500 MHz, DMSO-d6) δ 8.97 (t, J = 5.7 Hz, 1 H), 8.62 (s, 2 H), 8.16 (s, 1 H), 8.01 (s, 2 H), 7.98 – 7.93 (m, 4 H),
7.79 (d, J = 8.2 Hz, 2 H), 7.61 (d, J = 8.5 Hz, 2 H), 7.37 (d, J = 1.5 Hz, 2 H), 7.18 (s, 1 H, NH4
+), 7.00 (d, J = 8.5 Hz, 2 H), 4.70 (s, 4 H), 4.57 (t, J = 5.2 Hz, 4 H), 4.49 (t, J = 5.3 Hz, 2 H), 4.47 – 4.40 (m, 6 H), 4.20 (t, J = 4.6 Hz, 2 H), 3.88 (t, J = 5.2 Hz, 4 H), 3.82 – 3.75 (m, 4 H), 3.61 – 3.43 (m, 22 H), 3.41 (t, J = 6.5 Hz, 4 H), 2.94 (t, J = 6.4 Hz, 4 H), 2.40 (dd, J = 8.3, 6.5 Hz, 4 H), 2.29 (s, 6 H), 2.12 – 2.03 (m, 4 H).
13C NMR (101 MHz, DMSO-d6) δ 195.1, 161.4, 159.3, 150.2, 148.2, 145.9, 144.3, 144.1,
137.7, 134.3, 132.7, 130.9, 129.1, 127.5, 126.0, 125.7, 123.5, 123.1, 122.3, 116.7,
114.3, 113.0, 110.5, 104.0, 70.0, 69.8, 69.6, 69.6, 69.5, 69.5, 68.9, 68.8, 68.7,
68.6, 67.3, 49.7, 49.3, 48.5, 48.0, 45.6, 35.3, 30.5, 28.2, 26.5.
HRMS-ESI: m/z [M + 2H + 2Na-2NH4]2+ calcd for (C69H98 N20O19S4): 825.2727; found: 825.2735; m/z [M + 1H + 3Na-2NH4]2+ calcd for (C69H98 N20O19S4): 836.2636; found: 836.2651; m/z [M + 4Na-2NH4]2+ calcd for (C69H98 N20O19S4): 847.2546; found: 847.2563; m/z [M + 2H + 1Na-2NH4]+ calcd for (C69H98 N20O19S4): 1627.5561; found: 1627.5535; m/z [M + H + 2Na-2NH4]+ calcd for (C69H98 N20O19S4): 1649.5381; found: 1649.5352; m/z [M + 3Na-2NH4]+ calcd for (C69H98 N20O19S4): 1671.5200; found: 1671.5183.
UV/Vis: λmax = 412 nm (H2O, ε = 18 645 M−1 · cm−1), 417 nm (DMSO).
Fluorescence: λmax = 671 nm (H2O), 652 nm (DMSO); Φfl = 2.2% (H2O), 20.5% (DMSO).
Funding Information
This project was generously funded by the Volkswagen Foundation (Az. 93 438).




