

Subscribe to RSS
DOI: 10.1055/a-1743-4534
Synthesis and Applications of Asymmetric Catalysis Using Chiral Ligands Containing Quinoline Motifs
Authors
The authors would like to thank the Department of Science and Technology, India (DST-Ref.No.: SB/FT/CS-117/2014), the Science and Engineering Research Board (SERB-Ref.No.: EEQ/2018/000574)-Ramanujan Fellowship, and the National Institute of Technology Puducherry, Karaikal, India for providing financial support. This research was supported by the SERB-DST Grant No. RJF/2020/000038. V.D. gratefully acknowledges the Ramanujan Fellowship. The author RD acknowledge ICT-IOC, Bhubaneswar for providing necessary support. Rambabu Dandela thanks DST-SERB for Ramanujan fellowship (SB/S2/RJN-075/2016), Core research grant (CRG/2018/000782) and ICT-IOC start-up grant. K.B.D. would like to acknowledge the support from the Management, Principal-Dr. S. Sumaya and the Director-Research & Industry-Institute Relations-Dr. M.S. Irfan Ahmed, Thassim Beevi Abdul Kader College for Women, Kilakarai, Ramanathapuram, Tamil Nadu, India.

Dedicated to Professor Benjamin List
Abstract
In the past decade, asymmetric synthesis of chiral ligands containing quinoline motifs, a family of natural products displaying a broad range of structural diversity and their metal complexes, have become the most significant methodology for the generation of enantiomerically pure compounds of biological and pharmaceutical interest. This review provides comprehensive insight on the plethora of nitrogen-based chiral ligands containing quinoline motifs and organocatalysts used in asymmetric synthesis. However, it is confined to the synthesis of quinoline-based chiral ligands and metal complexes, and their applications in asymmetric synthesis as homogeneous and heterogeneous catalysts.
1 Introduction
2 Synthesis of Chiral Ligands Containing Quinoline Motifs
2.1 Synthesis of Schiff Base Type Chiral Ligands
2.2 Synthesis of Oxazolinyl-Type Chiral Ligands
2.3 Synthesis of Chiral N,N-Type Ligands
2.4 Synthesis of Amine-Based Chiral Ligands
2.5 Synthesis of P,N-Type Chiral Ligands
2.6 Synthesis of Chiral N-Oxide and Nitrogen Ligands
3 Homogeneous Catalytic Asymmetric Reactions
3.1 Asymmetric Carbon–Carbon Bond Formation Reactions
3.2 Asymmetric Allylic Reactions
3.3 Asymmetric Cycloadditions
3.4 Asymmetric Carbene Insertions
3.5 Asymmetric Pinacol Couplings
3.6 Asymmetric Pudovik Reactions
3.7 Asymmetric Strecker Reactions
4 Heterogeneous Catalytic Asymmetric Reactions
4.1 Asymmetric Cyclopropanation of Olefins
4.2 Asymmetric Heck Reactions
4.3 Asymmetric Hydrogenations
4.4 Asymmetric Hydroformylation of Styrene
4.5 Asymmetric Dialkoxylation of 2-Propenylphenols
4.6 Asymmetric Cascade Cyclizations
4.7 Asymmetric Allylic Alkylations
4.8 Asymmetric Alkylation of β-Keto Esters
4.9 Asymmetric C–H Bond Arylation Reactions
4.10 Intramolecular Aerobic Oxidative Amination of Alkenes
4.11 Asymmetric Oxidative Hydroboration of Alkenes
5 Conclusions
Key words
chiral ligands - catalysis - asymmetric synthesis - nitrogen heterocycles - quinoline motifs - organometallicsBiographical Sketches

Dr. Vasudevan Dhayalan obtained his MSc in organic chemistry (2005) and his PhD in organic chemistry (2011) at the University of Madras, Chennai, India. Then he received post-doctoral research experience with Prof. Masahiko Hayashi, Kobe University, Kobe, Japan, Prof. Paul Knochel, Ludwig-Maximilians-University, Munich, Germany, and Prof. Anat Milo, Ben-Gurion University of the Negev, Beer Sheva, Israel. He is a recipient of a PBC Outstanding Postdoctoral Research Fellowship. Recently, he was awarded a Ramanujan Fellowship from the Science and Engineering Research Board (SERB), India. Currently, he is working as a Ramanujan Fellow (Assistant Professor) at the National Institute of Technology Puducherry, Karaikal, India.

Dr. Rambabu Dandela obtained his MSc (organic chemistry) from Kakatiya University (2002–2004) and then worked as a research chemist at Matrix Laboratories, Hyderabad (2004–2008). He obtained his PhD from Dr. Reddy’s Institute of Life Sciences, University of Hyderabad campus, in 2013.Then he moved to Ben-Gurion University of the Negev as a PBC Outstanding Postdoctoral Research Fellow to work with Prof. Michael M. Meijer (2013–2017) where he was involved in the development of novel chemical probes for the study of quorum sensing processes in bacteria. In early 2017 he joined the CSIR-National Chemical Laboratory, Pune as a Ramanujan Faculty Fellow. In 2018, he became an Assistant Professor of Chemistry at the Institute of Chemical Technology, Indian Oil Odisha Campus, Bhubaneswar. His research interests lie at the interface of chemistry and biology with particular focus on structure-based drug design, bacterial signaling and polymorphism in pharmaceutical solids.

Dr. K. Bavya Devi is currently working as an Assistant Professor in the Department of Chemistry and Research Head at Thassim Beevi Abdul Kader College for Women, Kilakarai, Ramnad, Tamil Nadu. She received her MSc in chemistry from the University of Madras, India, in 2008. After that, she obtained her PhD in the Department of Chemistry, Anna University Chennai and carried out her research collaboratively with the Bhabha Atomic Research Centre (BARC) Mumbai. Dr. Bavya has received eight best paper awards in both national and international conferences. She was awarded a National Doctoral Fellowship (NDF) by the All India Council of Technical Education, New Delhi, India. Later, she joined Professor Mangal Roy’s group as a post-doctoral researcher in the Department of Metallurgical and Materials Engineering at the Indian Institute of Technology Kharagpur, India, in 2015. Her research interests are primarily focused on the development of new degradable materials that support bone regeneration.

Dr. Ragupathy Dhanusuraman received an MSc degree in chemistry from Bharathiar University, Coimbatore, India (2005). He completed his PhD in the Department of Chemistry, Kyungpook National University, South Korea (2010). Currently, he is working as an Assistant Professor in the Department of Chemistry, National Institute of Technology Puducherry, Karaikal, India. His research interests include organic polymers and the development of new nanomaterials for energy and electro-analytical applications.
Introduction
Quinoline, which is now one of the most important heterocyclic compounds, displaying a wide range of applications in pharmaceutical industries and organic synthesis, was first discovered by German chemist Friedlieb Ferdinand Runge in 1834, as a hygroscopic colorless liquid obtained by the distillation of coal tar.[1] The Friedländer annulation remains one of the simplest and most straightforward methods used in organic synthesis to access highly functionalized polysubstituted quinolines. This transformation is generally accomplished through the condensation of 2-aminoarylaldehydes or ketones with a ketone containing an active methylene group in the presence of acid or base.[2] [3] [4] Later, numerous methods were developed for the preparation of highly substituted quinoline and its derivatives.[5–11] Moreover, many quinoline derivatives exhibiting significant biological activities have been isolated from plants or systematically designed and synthesized.[12,13]
Quinolines and their derivatives have been labeled as ‘privileged scaffolds’ owing to their prevalent existence in natural and synthetic molecules that exhibit notable applications in pharmacological, agrochemical, and electronic industries (Figure [1]).[14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24]

Furthermore, quinoline analogues display antimalarial,[25] [26] [27] anti-inflammatory, antitumor, antibacterial, antiviral,[28] anticonvulsant,[29] and cardiovascular[30] activities.[31] Numerous studies on enantioselective catalysis have focused on the development of novel chiral ligands for use in organo- and transition-metal-catalyzed asymmetric reactions.[32] [33] [34] [35] [36] [37] [38] [39] [40] [41] Recently, Benjamin List and David MacMillan have clearly demonstrated the significance of organocatalyst in asymmetric synthesis.[41] Given the broad structural diversity of bicyclic nitrogen heterocycles, these recent Nobel Laureates have demonstrated the ability of these compounds to catalyze a range of chemical transformations. The ease with which many such quinoline (benzo[b]pyridine) ring systems can be synthetically modified within chiral scaffolds[42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] can lead to the discovery of new enantioselective processes for the synthesis of highly challenging chiral products with interesting applications in biology.
2
Synthesis of Chiral Ligands Containing Quinoline Motifs
2.1Synthesis of Schiff Base Type Chiral Ligands
In 2008, Hayashi and co-workers reported the preparation of the N,N,P-ligands. The N,N,P-tridentate Schiff base ligands 6a,b were prepared from chiral amino alcohols 1 in five steps with high yields (Scheme [1]). The synthetic pathway started from chiral amino alcohols 1, NH and OH-tosylation of which, followed by treatment with potassium hydroxide (KOH) gave the anticipated substituted aziridines 3a,b. The obtained amine-protected aziridines 3 were treated with KPPh2 to afford the corresponding N-tosylated amino phosphines 4. Simple condensation of 2-quinolinecarboxaldehyde with these N-H free amino phosphines 5 gave the expected N,N,P-tridentate chiral Schiff bases 6a,b in good yields. These N,N,P-tridentate Schiff base ligands were used quinoline-based asymmetric catalysts in organic synthesis, such as 1,4-addition of R2Zn to α,β-unsaturated ketones.

In the same year, Hayashi and co-workers reported the preparation of a library of chiral Schiff base ligands 11a–f (Scheme [2]). Chiral imines were readily prepared by condensation of aldehydes 7 or ketones 9 with chiral amines. The keto-imine chiral Schiff bases, 11a–f were prepared by two different methods. In method 1, the addition of a Grignard reagent[69a] to 2-quinolylaldehyde 7 produced the desired alcohol 8 in good yields; the obtained secondary alcohol was then effectively converted into the corresponding ketones 9 via a radical oxidation process. In method 2, treatment of 2-quinoline cyanide 10 with a Grignard reagent furnished the required ketones 9. Finally, condensation of ketones 9 with chiral amino alcohols in the presence of TiCl4 and Et3N gave the corresponding chiral ligands 11a–f (Scheme [2]). These N,N-bidentate Schiff base ligands were applied to the allylic oxidation of olefins.

In 2004, Suga and co-workers investigated the synthesis of diamine type chiral Schiff base ligands 14a–f. Aldimine type chiral Schiff bases were synthesized by simple condensation of substituted 2/8-quinolylaldehyde 13a,b with chiral 1,1′-binaphthyldiamine 12 in benzene under reflux (Scheme [3]). These binaphthyldiimine Schiff base ligands were found to be widely applicable to various 1,3-dipolar cycloaddition reactions and Diels–Alder reactions.

Eddine and co-workers demonstrated the preparation of iminium salt 17 via halogen–metal exchange reaction of (R)-2-(sec-butoxy)bromonaphthalene (15) with n-BuLi at low temperature to furnish the aryl lithium species 16, which, upon subsequent nucleophilic addition to 8-cyanoquinoline followed by quenching with methyl iodide (Scheme [4]), furnished the corresponding chiral N-methyl-1-(8-quinolinyl)-1-(2-(R)-sec-butoxynaphthyl)-methylenimime ligand 17 (Scheme [4]). These keto-imine type chiral Schiff base ligands were examined in effective phase-transfer catalyzed asymmetric alkylation reactions.

2.2
Synthesis of Oxazolinyl-Type Chiral Ligands
In 1999, Chelucci and co-workers designed a simple synthesis of chiral oxazolinylquinoline type ligands 21 and 22. Oxidation of quinoline 18 with 3-chloroperbenzoic acid (m-CPBA) in DCM for 2 h and then treatment of the obtained N-oxide with 2.0 equivalents of KCN and PhCOCl in CH3CN/MeOH at room temperature for 24 h, produced the corresponding compound 19. Subsequent treatment with 2-cyanoquinoline 19 and chlorobenzene under reflux with the addition of a suitable chiral amino alcohol 20a in the presence of ZnCl2 produced the corresponding oxazolinylquinoline type chiral ligands 21 and 22 in yields of 10–99% (Scheme [5]). In a similar manner, chiral quinoline ligands 24a–c were synthesized from 2-cyanomethylquinoline 23 and the corresponding amino alcohol 20a, mediated by ZnCl2 under reflux conditions (Scheme [5]).

Chelucci (2000) and co-workers expanded the library of quinoline ligands, by synthesizing chiral oxazolines 28a–e, employing an amide–mesylate–oxazoline reaction sequence (Scheme [6]). Thus, 8-quinolyl carboxylic ester 25 was heated in toluene under reflux with chiral amino alcohol 26a in the presence of potassium cyanide to afford the corresponding amide derivates 27 in quantitative yields. Finally, the quinolyloxazolines 28a–e were obtained by the reaction of amino alcohol 27 with methane sulfonyl chloride (MeSO2Cl) and Et3N in DCM solvent. The obtained chiral ligands 28a–e were air-sensitive and, upon standing at 25 °C, underwent a ring opening reaction to furnish the amide by-products. The chiral ligand 28d was treated with metal salts such as Cu(OTf)2 or PdCl2 to give the corresponding oxazoline transition-metal complexes 29. These chiral quinolyloxazoline ligands were studied in Friedel–Crafts alkylation, cyclopropanation of olefins, cascade intramolecular cyclization reactions, dialkoxylation of 2-alkenes, intramolecular aerobic oxidative amination, and allylic alkylation.

Muller and co-workers (2000) reported an innovative synthesis of oxazolinyl ligands 31 containing a hydroxyl group and silyl group 32 (Scheme [7]). The central chiral ligand was prepared by one-pot cyclization of 8-cyanoquinoline and chiral amino benzyl alcohol 26b at 110 °C in the presence of ethylene glycol. The obtained oxazolinyl-OH ligand 31 was protected using TBDPSCl in the presence of imidazole at room temperature for 24 h. Likewise, the benzyl protected oxazolinyl ligand 33 was prepared.

Ahn and co-workers (1999) designed and synthesized 8-diarylphosphino-2-oxazolinylquinoline type chiral ligands 38a–c starting from 35. 2-Cyano-8-hydroxyquinoline precursor 35, synthesized from 8-hydroxyquinoline 34 according to the literature,[75] was used for the preparation of the target chiral ligands. The authors reported that ZnCl2-catalyzed oxazolinyl ring formation furnished better yields when the quinoline-alcohol group was converted into the corresponding aryl triflate 36; otherwise, in the presence of the quinoline free OH group, oxazolinyl ligand derivatives 37 were obtained in lower yields. The condensation of l-valinol 20b and aryl cyanide 36 in the presence of ZnCl2 (10 mol%) with PhCl as a solvent, afforded the resulting oxazolines 37a–c in good yields. Introduction of the diphenylphosphino group (PPh2) was accomplished by Ni-catalyzed C–P coupling. Thus, reaction of the oxazoline derivatives 37 with diphenylphosphine in the presence of NiCl2(dppe) (10 mol%) and 1,4-diazabicyclo[2.2.2]octane (DABCO, 2 equiv) in DMF at 80 °C afforded the subsequent N,N,P-ligands 38a–c in moderate isolated yields. When the coupling reaction was carried out at 100 °C or above, as previously reported, the reaction yield was diminished (Scheme [8]).

2.3
Synthesis of Chiral N,N-Type Ligands
Bolm and co-workers prepared a wide range of quinoline-based C 1-symmetric chiral monosulfoximine derivatives 41a–l, in which the second donor nitrogen atom is in a quinolinyl aromatic ring, by Pd(OAc)2-catalyzed N-arylation of optically pure sulfoximines 39 with the corresponding 8-bromoquinoline derivatives 40. The chiral sulfoximine substrate scope is summarized in Scheme [9].

Several N,N-bidentate type chiral quinoline derivatives have been prepared from the corresponding ketones, as reported by Chelucci and co-workers in 2000. Chiral ligands 45a–c were prepared by the reaction of quinoline ketone 42 with vinyl ketone 43 (Scheme [10]) to produce the desired chiral quinoline intermediate 44, which was subsequently deprotonated with LDA at –78 °C and then treated with alkyl or benzyl iodide to give the corresponding alkylated ligands 45a–c.

Quinoline analogues 48a–c were successfully synthesized from methyl ketones 46 under similar reaction conditions (Scheme [10]) using LDA and alkyl halides.
A new class of chiral ligands containing the quinoline moiety 51a–d was developed by Yamamoto and co-workers in 2004. The coupling reaction of bis-aryl iodo compound 49 with quinoline derivatives 50 in the presence of LDA and BBr3 furnished the required bis quinoline compounds 51a–d (Scheme [11]). Chiral ligands 51a–d were then treated with Et2AlCl or CrCl2 to give the corresponding tethered bis(8-quinolinato) (TBOx) aluminum complexes 52a–d in good yields. These N,N-quinoline ligands were applied in pinacol couplings, the Pudovik reaction, hydrogenation of ketones and allylic alkylations.

2.4
Synthesis of Amine-Based Chiral Ligands
In 2007, Romanelli and co-workers reported a series of quinoline ligands 57 prepared by the alkylation of alcohol 53 in the presence of TsCl (1.2 equiv) and pyridine at room temperature. Subsequent alkylation of 6-hydroxyquinoline 55 with Ts-ester 54 in the presence NaH (2 equiv) in DMF at 80 ° for 4 h was followed by reduction with LAH and then MeI was added to furnish the required chiral amine salt (R)-57 in high yield (Scheme [12]).

Kwong et al. introduced a novel synthesis of bisamide ligand-containing quinolines, whose asymmetric synthesis started from condensation of cyclohexyl diamine 59 with heterocyclic aldehydes 58 and 61 to give amide-based unsymmetrical ligands 62 and symmetrical ligands 60 in good yields (Scheme [13]).

Judeh and co-workers described a series of quinoline ligand derivatives 68a–m, whose synthesis starts from simple condensation of phenylethylamine 63 with diethyl oxalate in ethanol to give compound 64 in high yield (Scheme [14]). Then, rac-65 was synthesized under double Bischler–Napieralski conditions. Bis-amide 64 was then reacted with polyphosphoric acid (PPA) at 190 °C for 12 h to furnished the target compound rac-65 in 86% yield. Reaction of compound 65 with a stoichiometric amount of enantiopure (S)-(–)-α-methylbenzyl isocyanate furnished the diastereomeric urea analogues 66a and 66a′ in excellent yield. When a solution of 66a or 66a′ was treated with n-BuONa in warm n-BuOH (Scheme [14]), the cleaved products (+)-67a and (–)-67a′, were obtained in up to 61% yield and 99% ee. Fortunately, one of the products could be recrystallized from ethanol and gave a very high enantiomeric excess >99%.

Various alkyl groups were introduced by reaction of (+)-67 with alkyl halides in the presence of K2CO3 with CH3CN as a solvent at 50 °C. Likewise, compound (+)-67 reacted with 1 equivalent of isocyanates and thioisocyanates in DCM at room temperature to give the target products 68a–m in excellent yields (Scheme [14]).
Yus and co-workers studied a practical method for the preparation of camphor sulfonamide-based quinoline ligands 71a,b. Their synthesis started from cyclohexyldiamine 59 by reaction with arylsulfonyl chloride in two steps, followed by treatment with camphor sulfonyl methyl chloride 70. Friedlander annulation in the presence of ruthenium chloride as a catalyst then furnished the expected camphor sulfonamide-based quinoline ligands 71a,b in moderate yields (Scheme [15]).

Felluga et al. efficiently synthesized the enantiopure amine-based ligands 75. Baker’s yeast mediated reduction of methyl ketone 72 afforded alcohol (S)-73. However, the required chiral alcohol (S)-73 could also be obtained by a kinetic resolution approach. Thus, azide precursors (R)-74 were obtained in good yield from the benzyl alcohol in the presence of DPPA/DBU and reduction with triphenylphosphine (Ph3P) led to the desired amine ligands 75 with high enantioselectivity (Scheme [16]).

Osmium metal complexes 77 and 78 were prepared by treatment of [OsCl2(PPh3)3] with (S,R)-Josiphos (1.2 equiv) in mesitylene at 110 °C for 2 h to give an uncharacterized mixture of products, which was then reacted with 2-aminomethylbenzo[h]quinoline 75 (1.4 equiv) in the presence of triethylamine (Et3N) at 140 °C for 24 h to furnish the corresponding coordination metal complexes 77 and 78 in good yields (Scheme [16]). These amine-based ligands were studied in catalytic applications such as 1,2-addition of organozinc reagents to substituted aldehydes, 1,4-addition of Grignard reagents (R1MgX) to cyclic enones, allylic alkylations, and C−H bond arylation reactions
2.5
Synthesis of P,N-Type Chiral Ligands
The efficient synthesis of QUIPHOS type chiral ligands 81a–h by the reaction of phosphane 79 and pyrrolidine 80 followed by addition of hydroxyquinoline 34 (method 1, Scheme [17]) afforded the desired ligands 81a–h in moderate to good yields, as reported by Buono and co-workers. Applying a similar reaction protocol led to a wide range of P,N-quinoline–phosphine ligand derivatives 83a–h; selected examples are shown in method 2, Scheme [17].

Quinoline-based chiral Pd complex 87 was effectively prepared via halogen–metal (Li–Br) exchange of heterocyclic bromo compound 84 and s-BuLi, followed by quenching with PCl(NMe2)2 to give quinoline–phosphine ligand 85. Reaction of P,N-ligand 85 with chiral amine 80 produced the corresponding chiral ligand 86 and this was treated with [PdCl2(CH3CN)2] in DCM to produce the desired N,N,P-Pd complex 87 in excellent yield (Scheme [18]).

The phosphonito, nitrogen ligand (R)-90 has been synthesized in a one-pot, two-step process (Scheme [19]). trans-Metalation of 8-bromoquinoline 84 with n-butyllithium (n-BuLi) and subsequent treatment with PCl(NEt2)2 to form phosphine compound 88, followed by the reaction with (R)-binaphthol 89 in toluene at reflux, furnished P,N-ligand (R)-90 in good yield. This ligand was reacted with Pd, Pt and Rh complexes to furnish the desired metal complexes 91–93 in good yields.

In 2000, Faraone and Leitner introduced the enantioselective synthesis of phosphane/phosphoramidite ligands 96a and 96a′ in a one-pot procedure from readily available 8-biarylphosphinoquinoline 94 by nucleophilic addition of organometallic lithium reagents and direct quenching with PCl3 to obtain P,N-ligand 95, followed by addition to chiral 1,1′-bi-2-naphthol 89 in the presence of Et3N. Under the same reaction conditions, a 1:1 mixture of diastereomers containing 2-substituted quinoline ligands 99a–d and 99a′–d′ was obtained from phosphinoquinoline 94. Selected examples are illustrated in Scheme [20].

Knochel and co-workers examined the synthesis of P,N-ligands 104 from commercially available starting materials. Treatment of (+)-camphor 100 with Tf2NPh in THF at 0 °C produced the desired compound 101 in 90% yield (Scheme [21]). The chiral camphor triflate 101 efficiently underwent a Pd-catalyzed Negishi cross-coupling reaction with the quinoline organozinc reagent,[88`] [b] [c] [d] affording the desired 2-alkenylquinoline 102 in acceptable yield. Subsequent hydrophosphination with Ph2P(O)H, in the presence of a catalytic amount of t-BuOK (20 mol%) in DMSO provided phosphine oxide 103 (Scheme [21]). Reduction of compound 103 was accomplished in the presence of HSiCl3 and Et3N in toluene at reflux, to generate the chiral aminophosphine 104 in good yield.

Chiral Ir complex 105 was synthesized by reaction of [Ir(cod)Cl]2 and P,N-ligand 104 in DCM at reflux. After treatment with NaBArF in a biphasic DCM–H2O system, the subsequent orange colored salt 105 was obtained after chromatographic purification. The iridium chiral metal complexes were stable towards moisture and oxygen.
Jiang et al. have designed and synthesized a series of phosphine–quinoline ligands. Their synthetic protocol began from optically pure paracyclophane 106. Hence, treatment of (Rp )-106 with n-butyllithium (n-BuLi) followed by successive addition to 2-quinolinylcarboxaldehyde, produced two diastereoisomers, (Sp ,S)-107a′ and (Sp ,R)-107a that could be readily separated by flash column chromatography (Scheme [22]). Modifying (Sp,S)-107a′ and (Sp,R)-107a by silylation in the presence of TBSOTf and lutidine as a base produced (Sp,S)-108a′ in 98% yield and (Sp,R)-108a in 97% yield, respectively.

Ruzzicon et al. investigated the valuable synthesis of P,N-bidentate planar chiral ligands 111 and 114. Deprotonation of methyl compound 109 with n-BuLi at 0 °C, involved the 2-methyl quinoline, giving the 2-methyllithium intermediate, exclusively. The borane complex (R)-110 was achieved in high yield, by the reaction of Ph2PCl with BH3·OMe2 (Scheme [23]). The subsequent air-stable borane complex (R)-110 was treated with DABCO, to obtain the expected phosphine (R)-111. On the other hand, bromo-compound 113 was prepared from alcohol 112 by treating with CBr4 and PPh3 in Et2O at 25 °C and successfully underwent nucleophilic substitution with lithium (diphenylphosphine)methylborane complex, followed by treatment with DABCO, providing the corresponding P,N-chiral ligand (R)-114 in 70% overall yield (Scheme [23]).


Brown et al. focused on the synthesis of chiral ligands 121a–g (QUINAP). Boronic acid 116 underwent smooth cross-coupling with aryl chloride 115 in the presence of 3 mol% Pd(PPh3)4 and Na2CO3 in DME to give carbon–carbon coupled product 117 in 96% yield. Cleavage of the methyl group from aryl methyl ether 117 with boron tribromide (BBr3) gave the required phenol analogue 118, which was further converted into the triflate 119 (Scheme [24]). Finally, palladium-catalyzed cross-coupling of triflate 119 with diphenylphosphine oxide gave the phosphine oxide 120. Subsequently, compound 120 was reduced to the phosphine ligand 121 with HSiCl3 and Et3N in 84% yield.
Finally, the racemic ligand 121 was reacted with the chiral palladacycle 122 to form diastereomers, from which the desired enantiopure R or S ligands 121a–g were obtained in good yields after fractional recrystallization and ligand decomplexation (Scheme [24]).

Furthermore, the same group developed a method for the preparation of benzo ring fused isoquinoline and indole-based chiral P,N-ligands 128 and 134 (Scheme [25] and Scheme [26]).


Ding and co-workers have synthesized spiro-based P,N-ligand 146 through a sequence of reactions as shown in Scheme [27]. Nucleophilic addition of compound 136, to a ketal derivative 137 generated a protected spiro-diketone 138. Then Friedländer condensation of 139 with 2-amino benzaldehyde 140 in the presence of KOH and EtOH furnished the polycyclic quinoline 141 in 70% yield. Selective deprotection of compound 141 in aq. TFA at room temperature for 1 h furnished the corresponding spiro-ketone 142 in excellent yield. Subsequent treatment of spiro-compound 142 with LiHMDS, followed by addition of PhNTf2, gave enol triflate 143 in 96% yield. Next, the coupling reaction of compound 143 with Ph2P(O)H in the presence of Pd catalyst afforded racemic phosphine oxide 144 in 85% yield, which was readily resolved by chiral HPLC to give both enantiomers in enantiomerically pure form.
The resulting chiral phosphine oxide 145 was simply reduced with HSiCl3 in the presence of pyridine, affording the required chiral nitrogen ligand (S)-146 in moderate yield (Scheme [27]). The reaction of nitrogen based P,N-ligand 146 with [Ir(cod)-Cl]2 in DCM followed by addition of NaBArF after counter-anion exchange gave the corresponding desired Ir metal complex (+)-147 in 87% yield.

Multi-step synthesis of silyl substituted chiral quinolinyl phosphane ligands 153a–c has been achieved by Pfaltz and co-workers. In the initial step, hydroxylation of compound 148 using a metal catalyst gave the corresponding 1,2-diol 149 in moderate yield and high enantiomeric excess on a gram scale (46% yield, 94% ee). Selective tosylation of the primary alcohol 149 in the presence of TsCl with pyridine, followed by silylation of benzyl alcohol 150 using TBDMSCl and imidazole generated enantiomerically pure compound 151 after recrystallization.
Next, sulfonate 151 was treated with LiPPh2·BH3 at –78 °C to furnish the phosphine-protected ligand 152 in good yield (Scheme [28]). Finally, the P–B bond was successfully cleaved using diethylamine to afford the desired P,N-ligands 153a–c in good yield.
Additionally, Ir-based transition-metal complexes 154a–c were produced from N-heteroaryl phosphane derivatives 153a–c. Warming a DCM solution of the requisite organocatalysts 153 in the presence of [Ir(cod)Cl]2 (0.5 equiv) for 2 h at 30–40 °C followed by counter-ion exchange with NaBArF (1.6 equiv), provided the metal complexes as orange solids. These types of metal complexes are generally stable to air and moisture, and are simply purified by flash column chromatography on silica gel (Scheme [28]).

Chelucci et al. reported a new class of bidentate ligands 160, 164 and 168 that were synthesized from the corresponding starting materials (+)-nopinone, (+)-camphor and 5-androst-2-en-17-one. The direct lithiation of compound 155 with t-BuLi at low temperature and then quenching with electrophile DMF affording coupled aldehyde 156. The N-Boc aldehyde 156 thus obtained reacts with acyclic ketone 157 in the presence of t-BuOK at 25 °C, leading to 159 in good yield (Scheme [29]). Finally, treatment of compound 159 with LiPPh2 gave the desired acridine 160 in 82% yield. The same group used similar reaction conditions to prepare additional quinoline-based P,N-chelating chiral ligands 164 and 168 in good yields (Scheme [29]).
Wild and co-workers introduced an efficient method for the preparation of (R or S)-carbene ligands 173 (Scheme [30]). The reaction of halogenated quinoline 169 with Na(PMePh) in THF furnished the desired compound 170 in very good yield. This racemic product was resolved by crystallization of a pair of internally diastereoisomeric Pd(II) complexes (R,R)- and (R,S)-171a,a′ derived from the chelating ligand (R)-122. The resulting tertiary phosphine (R)- and (S)-172 was accessed by treatment with H2SO4 and LiCl (Scheme [30]). Finally square-planar palladium complexes (R)- and (S)-172 were successfully converted into the optically pure enantiomers (S)- and (R)-173 with aq. KCN and DCM/H2O in a biphasic reaction medium.

These quinoline-based P,N-ligands were broadly applied as asymmetric catalysts in cyclopropanation of olefins, Heck reactions, hydrogenation of olefins, ketones and imines, hydroformylation, allylic alkylation, and oxidative hydroboration.
2.6
Synthesis of Chiral N-Oxide and Nitrogen Ligands
Martinez et al. developed an efficient method for the preparation of camphor sulfonamide-based quinoline chiral ligands and their N-oxide derivatives. These chiral amine ligands 174a–c (C 2-symmetry) were prepared by the addition of camphorsulfonyl chloride 70 to 1,2-cyclohexanodiamine 59 and, without additional purification, the resulting intermediates were treated with an aminobenzyl alcohol (Scheme [31]) to afford the desired camphor sulfonamide-based quinoline ligands 174a–c in high yields. The quinoline N-dioxide ligands 175a–c were simply synthesized from the corresponding ligands 174a–c (C 2-symmetry) by oxidation with mCPBA in DCM at 0 °C. The resulting amine type N-dioxide ligands 175a–c were formed in reasonable yields and were typically stable enough to be purified by flash column chromatography.


Nakajima et al. successfully developed a protocol for the synthesis of C 2-symmetric 2,2′-biquinoline N,N′-dioxide (R or S)-178 and 1,1′-biisoquinoline N,N′-dioxide (R or S)-182 (Scheme [32]). The racemic compound 177 was prepared by mCPBA oxidation of 3,3′- dimethyl-2,2′-biquinoline 176, and the product was resolved through a hydrogen-bonding complex with (S)- or (R)-binaphthol to afford desired chiral compounds (R)-178a and (S)-178a′ (Scheme [32]). The enantiomerically pure ligand 1,1′-biisoquinoline N,N′-dioxide (S)-182 was prepared by preparative chiral HPLC from racemic compound 181, which was in turn synthesized by N-oxidation of 1,10-bisisoquinoline 180 using H2O2.
The racemic compound 117 was prepared from 1-chloro isoquinoline 115 via Suzuki cross-coupling reaction in the presence of boronic acid 116. Racemic 117 was further reacted with mCPBA, and was resolved via a complex with (S)-binaphthol to give the required chiral compounds (R)-183.
The ligands (–)-185 and (+)-186 were obtained by resolution of rac-184 with d and l-dibenzoyltartaric acid, respectively. The absolute configuration of chiral ligand (S)-186 was determined by single-crystal X-ray analysis (Scheme [32]). Quinoline-based N-oxide ligands were studied in various asymmetric catalytic reactions such as 1,4-addition and Michael addition reactions, allylation of aromatic and heteroaromatic aldehydes, and Strecker reactions.
Finally, in this section, Meyers et al. established the synthesis of chiral naphthylquinoline ligands 188 and 189. Addition of naphthyllithium (1.1 equiv) to quinoline oxazoline 187 in THF at –78 °C for 2–3 h followed by oxidation with dichlorodicyanoquinone (DDQ) in THF at –78 °C gave 1-naphthyl-4-quinoline (S)-188a,b in good yield. A similar process using 187 and arylmagnesium reagents[97a] followed by oxidation with DDQ (THF, –78 °C) gave the biaryl compounds (R)-189a,b in high yields (Scheme [33]).

3
Homogeneous Catalytic Asymmetric Reactions
3.1Asymmetric Carbon–Carbon Bond-Formation Reactions
Catalytic asymmetric C–C bond-forming reactions provide one of the most efficient methods to synthesize chiral molecules, and a range of pyridine and quinoline-based chiral catalysts have been developed in the past two decades, finding a wide range of applications.[14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31]
3.1.1Asymmetric Addition of Dialkylzinc to Aldehydes
In 2008 Cozzi, Yus, Ramón and co-workers described the preparation of camphor sulfonamide-based quinoline ligands. This type of chiral quinoline ligands has been used for the synthesis of trisubstituted chiral alcohols. The enantioselective 1,2-addition of organozinc reagents to substituted aldehydes 190, provides alcohols 191 with high enantioselectivities (up to 96% ee) with either aromatic or aliphatic substrates (Scheme [34]). These reactions were carried out using 10 mol% chiral amine ligand 71a organozinc reagent (2.4 equiv) and 1.1 equivalents of Ti(O-i-Pr)4.

In 2010, Judeh and co-workers synthesized constrained chiral C 1-symmetric 1,10-bisisoquinoline ligands. The consequences of their geometrical conformations were found to have a significant effect on the catalytic asymmetric addition of diethylzinc to aromatic aldehydes 190. To study the reaction scope and limitations of ligand (+)-67a′, several aromatic aldehydes having electron-donating and electron-withdrawing substituents were examined under the optimized reaction conditions. In general, this protocol produced excellent yields and high enantioselectivities of the secondary alcohols 192 (Scheme [35]).

3.1.2
Asymmetric 1,4-Additions of Dialkylzincs to Enones
Buono and co-workers investigated the use of a copper catalyst involving QUIPHOS 81a–h as a chiral ligand. This system was applied to the 1,4-addition of Et2Zn to α,β-unsaturated cyclic ketones. Notably, additives such as water or zinc hydroxide had a significant effect, leading to an improved enantiomeric excess from 7 to 61% ee in this enantioselective 1,4-addition system (Scheme [36]).
Faraone and co-workers examined the Cu(II)-catalyzed asymmetric 1,4-addition of diethylzinc to 2-cyclohexen-1-one, in the presence of a catalytic amount of chiral ligands 96a and 83h with appropriate metal salts. The 1,4-adducts were formed with enantioselectivities up to 70% ee with BINAPHOSHQUIN 96a (Scheme [36]).

Later, Hayashi and co-workers developed mild and effective methods for the synthesis of chiral alcohols with excellent enantioselectivity. The copper-catalyzed enantioselective conjugate 1,4-addition of dialkylzinc reagents to α,β-unsaturated cyclic ketones 193 with catalytic amounts (0.2 mol%) of Cu(OTf)2 and 0.25 mol% of one of the N,N,P-tridentate Schiff base ligands 6a,b gave cyclic ketone adducts 194 in up to 99% ee in good yield (Scheme [36]). The impact on the enantioselectivity and reactivity of many other variables, such as the nature of the metal catalyst, ligands, and ligand/catalyst loading involved were also examined in detail and the results obtained are summarized in Scheme [36].
Moreover, Hayashi and co-workers further expanded the scope of the 1,4-addition reaction to access disubstituted ketones via copper-catalyzed 1,4-addition of dialkylzincs to α,β-unsaturated ketones. The reactive zinc enolate intermediates were trapped efficiently with reactive allyl iodides to afford the corresponding disubstituted ketones 195 with excellent diastereo- and enantioselectivity. The desired 1,4-addition reactions were performed using 1 mol% Cu(OTf)2 and 1.5 mol% Schiff base ligand 6a,b. The results obtained are summarized in Scheme [37].

3.1.3
Asymmetric Conjugate Addition of Grignard Reagents to Enones
Highly constrained C 1-1,10-bisisoquinoline chiral ligands (+)/(–)-67 were examined in the enantioselective 1,4-addition of Grignard reagents (R1MgX) to cyclic enones 193. The desired 1,4-adducts 194 were obtained in very good yields but with low enantioselectivity (up to 35% ee) (Scheme [38]).

3.1.4
Asymmetric Conjugate 1,4-Addition of Thiols to Cyclic Enones
Nakajima and co-workers examined the enantioselective conjugate nucleophilic 1,4-addition of thiols to enones under mild reaction condition, leading to the corresponding sulfides 196 with moderate enantioselectivities (up to 78% ee). This protocol provided the first example of using a cadmium complex in an asymmetric thiol 1,4-addition reaction (Scheme [39]).

3.1.5
Asymmetric Michael Addition Reaction
In 2003, Nakajima et al. studied the catalytic, enantioselective Michael addition of β-keto esters to α,β-unsaturated carbonyl compounds using a chiral biquinoline N,N′-dioxide–Sc(OTf)3 (R)-178a complex as catalyst. The Michael adducts 198 were produced in good yields with moderate enantioselectivities (up to 84% ee) (Scheme [40]). Electron-donating indanone substrates 197 were tested using 5 mol% quinoline N,N′-dioxide ligand–Sc(OTf)3.

3.1.6
Asymmetric Friedel–Crafts Alkylation
Zhou and co-workers (2006) investigated an efficient asymmetric Friedel–Crafts alkylation of free N–H indoles 199 with nitro compound 200 catalyzed by Zn(OTf)2-oxazoline complexes 28e. The nitroindole 201 was prepared in good yield, but very low enantioselectivities (up to 9% ee) were observed in the presence of 12 mol% chiral ligand 28e and 10 mol% Zn(OTf)2 at 0 °C for 20 h (Scheme [41]).

3.2
Asymmetric Allylic Reactions
3.2.1Asymmetric Allylic Oxidation
In 2008 Hayashi and co-workers studied the copper (I)-catalyzed enantioselective allylic oxidation of several cyclic olefins with tert-butyl perbenzoate (PhCO3But) enabled by N,N-bidentate Schiff base ligands 11a, which were effective in conferring high reactivity and moderate-to-good enantioselectivity (up to 84% ee). The authors examined the allylic oxidation of numerous cyclic olefins 202 using a Cu(CH3CN)4PF6 and Schiff base ligand system. The results, summarized in Scheme [42], were obtained using a catalytic amount of chiral N,N-bidentate ligand 11a.

Later, in 2009, Hayashi and co-workers developed an enantioselective desymmetrization by allylic oxidation of 4,5-epoxycyclohex-1-ene 205 in the presence of 3 mol% of chiral N,N-bidentate Schiff base ligand 11a and 2.5 mol% of Cu(CH3CN)4PF6 to afford phenyl epoxide 206 in 84% ee, which was improved to >99% ee after derivatization with 4-nitro benzoyl chloride and recrystallization to give the corresponding nitroaryl epoxide derivatives 207 (Scheme [43]).

3.2.2
Asymmetric Allylation of Aldehydes with Allylchlorosilanes
In 2002, Malkov, Koćovský and co-workers developed the Sakurai–Hosomi-type allylation of aromatic aldehydes 190 catalyzed by C 2-symmetric 2,2′-biquinoline N,N′-dioxide (S)-178, leading to the corresponding chiral alcohol 209 in good enantioselectivities (up to 88% ee) and 85% reaction yield (Scheme [44])

Later, the same group (2003) revealed that the addition of allyltrichlorosilane 208 to aromatic aldehyde 190 in the presence of quinoline N-oxide ligand (R)-183 (5 mol%) at –40 °C in DCM for 0.5–12 h, produced the corresponding alcohol derivatives 209 with 5–96% ee and good yields. The aldehyde substrate scope is summarized in Scheme [45].

3.2.3
Asymmetric Phase-Transfer Allylic Alkylation
Later, in 2008, Eddine and co-workers reported the phase-transfer-catalyzed asymmetric alkylation of ester 210 with allyl bromide in the presence of 10 mol% of chiral N,N-bidentate Schiff base salt 17 with the use of NaOH, affording the desired compound 211 in good yield and very low enantioselectivity (Scheme [46]).

3.3
Asymmetric Cycloadditions
3.3.1Asymmetric Diels–Alder Reactions
Buono and co-workers (1998) reported the asymmetric Diels–Alder reaction catalyzed by copper-phosphene complexes. The nitrogen-based copper(II) catalyst was prepared by mixing Cu(OTf)2 and chiral quinolinephosphine ligand 81a in DCM and further used in the Diels–Alder reaction of 3-acryloyloxazolidin-2-one 213 with cyclopentadiene 212, leading to the corresponding amide product 214 in excellent yields and remarkable enantioselectivities (up to 99%) (Scheme [47]).

Subsequently, in 2004, Suga et al. developed an efficient method for Ni(II)-catalyzed asymmetric Diels–Alder reactions of cyclopentadiene 212 and 3-alkenoyl-2-oxazolidinones 215 in the presence of the ligand BINIM-2QN 14a (Scheme [48]). Even loadings down to 1 mol% Ni(II) catalyst promoted Diels–Alder reactions with high conversions and enantioselectivities (endo-addition with up to 94% ee).

3.3.2
Asymmetric Hetero-Diels–Alder Reactions

Bolm et al. studied the first example of a copper-catalyzed hetero-Diels–Alder reaction of cyclohexa-1,3-diene (217) and keto ester 218 in the presence of 10 mol% Cu(OTf)2 and C 1-symmetric sulfoximine ligands 41a–l, leading to cycloadducts in good yields and high enantioselectivities (up to 96% ee) as shown in Scheme [49].
Asymmetric cycloaddition of nitrones 220 and 3-(2-alkenoyl)-2-thiazolidinethiones 221 using chiral binaphthyldiimine–Ni(II) complexes 14a–e to afford products in high exo-diastereoselectivities and enantioselectivities was reported by Suga et al. in 2005 (Scheme [50]).

3.3.3
Asymmetric 1,3-Dipolar Cycloaddition Reactions
Shi and co-workers reported chiral binaphthalenediimine-Ni(II) complex 14a as an active catalyst in the 1,3-dipolar cycloaddition reactions of azomethine ylides 223 and 1-phenyl-1H-pyrrole-2,5-dione 224 to give the corresponding adducts 225 in very low yields and poor enantiomeric excesses (up to 8%) in Scheme [51].

Furthermore, in 2011, Suga et al. demonstrated that BINIM–Ni(II) catalysts 14a–c were efficient for enantioselective 1,3-dipolar cycloaddition reactions between ethyl diazoacetate 226 and 3-acryloyl-2-oxazolidinones 227 to produce the corresponding adducts 228 in high yields and enantiomeric excesses (up to 93%) as shown in Scheme [52].

The same group had previously reported in 2007 the first example of highly enantioselective 1,3-dipolar cycloaddition reactions between azomethine imines 229 and 3-acryloyl-2-oxazolidinone 230 using 10 mol% of chiral BINIM–Ni(II) complex 14b (Scheme [53]).

3.4
Asymmetric Carbene Insertions
A simple and efficient method for Cu-catalyzed enantioselective C–H carbene insertion between methyl phenyldiazoacetate and THF in the presence of 2.2 mol% of chiral N,N-bidentate Schiff-base ligand 28e and 2.0 mol% of copper catalyst to afford the corresponding syn-product 234 in 45% ee, was reported by Fraile et al. in 2007. Copper salts such as Cu(OTf)2, CuBr2, Cu(OAc)2, CuCl, and CuSbF6 were examined to optimize the reaction and copper triflate furnished better results (Scheme [54]).

3.5
Asymmetric Pinacol Couplings
In 2004, Yamamoto and co-workers introduced a new class of chiral tetradentate ligand, TBOx 52a, as a catalyst for pinacol coupling. Chromium complex TBOxCrCl 235 was shown to be an efficient catalyst for the asymmetric pinacol coupling reactions of both functionalized aromatic and aliphatic aldehydes 190. With aromatic substrates, the catalyst system was shown to be quite insensitive to changes in steric effects on the substrates as well as to the presence of electron-donating and electron-withdrawing substituents on the aromatic ring, providing high enantiomeric excesses (up to 98%, Scheme [55]).

3.6
Asymmetric Pudovik Reactions
The same group in 2008 developed the catalytic enantioselective Pudovik reaction of aldehydes 190 and aldimines 239 with tethered bis(8-quinolinato) (TBOx) aluminum complexes 52a–d. α-Hydroxy- and α-aminophosphonates 238 and 240 were prepared in high yields and enantioselectivities (96–98% ee) using a low catalyst loading (1 mol%). This was a significant improvement over other catalysts in that they generally required higher catalyst loadings, typically >5 mol% and extended reaction times. The chiral ligand could be easily recovered in high purity after simple purification without loss in either reactivity or selectivity (Scheme [56]).

3.7
Asymmetric Strecker Reactions
Feng and co-workers (2003) investigated enantioselective Strecker reactions with trimethylsilyl cyanide (TMSCN) 242 and aryl imines 241 catalyzed by chiral N,N′-dioxide ligand 178. These chiral quinoline N,N-dioxide Lewis base promoters were effectively applied to the chiral synthesis of α-amino aryl nitrile analogues 243 with high enantioselectivities (up to 95% ee). Enantiomerically pure products (up to 99% ee) were subsequently obtained by recrystallization (Scheme [57]).

4
Heterogeneous Catalytic Asymmetric Reactions
Fraile, Mayoral and co-workers reported quinoline-based oxazoline ligands, a class of C 1-symmetric chiral ligands, in the enantioselective cyclopropanation of styrene (244) with ethyl diazoacetate 245 in DCM at 25 °C, which proceeded with excellent cis-selectivity (up to 65%). This result may be synthetically of interest, given that cis-cyclopropanes are generally difficult to obtain. The substrate scope is summarized in Scheme [58].

Asymmetric Cyclopropanation of Olefins
In 1998, Ahn and co-workers studied the Ru(II)-catalyzed intramolecular cyclopropanation of diazo-alkenes 247. The catalytic chiral system demonstrated good reactivity and stability, and produced high yields with moderate enantioselectivities (Scheme [59]).

4.2
Asymmetric Heck Reactions
In 2004 Pfaltz and co-workers described the generality and utility of ligands 153a,b in palladium-catalyzed enantioselective Heck reactions. The results are summarized in Scheme [60].

4.3
Asymmetric Hydrogenations
4.3.1Asymmetric Hydrogenation of Alkenes
P,N-Chiral iridium complexes 154a–c were efficiently applied to asymmetric hydrogenation of di-substituted alkenes 253 (Scheme [61], entries 1–3), resulting in up to 56% ee, as reported by Pfaltz and co-workers in 2004. The reactivities of the metal complexes are summarized in Scheme [61]. In general, phosphinites were excellent in terms of both enantioselectivity and reactivity. Additionally, in 2003, Knochel and co-workers demonstrated that ligand 105 mediated Ir-catalyzed asymmetric hydrogenation reactions of tri-substituted alkenes (Scheme [61], entries 4–6) leading to hydrogenated products with high enantioselectivity (up to 95% ee).

4.3.2 Asymmetric Hydrogenation of Ketones
In 2005, Leitner and co-workers developed a highly enantioselective ruthenium-catalyzed hydrogenation of aromatic ketones with (R a,S c)-QUINAPHOS 99a in the presence of substituted and non-substituted diamines as co-catalysts. The hydrogenation results obtained are summarized in Scheme [62].

Later, in 2010, Baratta et al. employed ruthenium metal complexes (MC) 255a–d and osmium complexes 77 and 78 in the presence of t-BuOK, to catalyze chemoselective asymmetric hydrogenation (5 atm H2) of aromatic and aliphatic ketones to give the desired chiral alcohols in high conversions and good selectivities (Scheme [62]).
4.3.3
Asymmetric Hydrogenation of Imines
In 2010, Ding and co-workers reported a chiral ligand bearing a spiro-scaffold-based Ir-complex 147 and successfully applied it in the enantioselective hydrogenation of aryl-imine 258, furnishing the corresponding chiral amine with enantioselectivities up to 58% ee (Scheme [63]).

4.4
Asymmetric Hydroformylation of Styrene
Rh-catalyzed asymmetric hydroformylation of styrene 260 in the presence of P,N-chiral quinoline ligand 99a was reported by Leitner and co-workers in 2007. The P,N-chiral Rh complexes were applied to asymmetric hydroformylations of mono-substituted alkenes to give the corresponding product 261 with up to 74% enantiomeric excess, with a linear aldehyde by-product 262 also being observed (Scheme [64]).

4.5
Asymmetric Dialkoxylation of 2-Propenylphenols
Sigman and co-workers (2007) successfully developed a direct O2-coupled Pd(II)-catalyzed enantioselective dialkoxylation of 2-alkenylphenols by using quinoline oxazoline ligands 21a–d (Scheme [65]). Pd(II)-catalyzed enantioselective dialkoxylation of 2-alkenylphenols 263, at room temperature for 24–72 h furnished the desired phenol 264 with enantioselectivities up to 92% ee.

4.6
Asymmetric Cascade Cyclizations
In 2009 Yang and co-workers reported the structurally tunable and an air-stable oxazoline 21c-Pd catalyst system for the highly enantioselective oxidative cascade intramolecular cyclization reaction of a variety of substituted bis-olefins 265, with excellent enantioselectivities (up to 98% ee), good yields and high diastereoselectivities (dr >24:1) (Scheme [66]).

4.7
Asymmetric Allylic Alkylations
Several chiral phosphine-quinoline ligand analogues were found to be good candidates for Pd-catalyzed asymmetric allylic alkylation reactions, as reported by Jiang et al. in 2008. Catalytic allylic alkylation has been demonstrated to be a powerful tool for stereoselective carbon–carbon bond-formation reactions in the presence of palladium-nitrogen ligand systems. Among many quinoline-based ligands designed for this chiral reaction, chiral bi- and tri-dentate type P,N-ligands have played a significant role owing to their electronic and steric parameters. The reactions were carried out using 1.0–6.4 mol% Pd catalyst and 2.5–12.8 mol% chiral quinoline ligand. The results from a range of ligands are summarized in Scheme [67]. Other protocols have been successfully examined for allylic alkylation reactions using various phosphine-quinoline based chiral ligands as outlined in Scheme [67].[72] [74] [77] [84] [89] [90] [94] [110]

Trost and co-workers (2002) investigated Mo-catalyzed enantioselective allylic alkylations with sodium dimethyl malonate in the presence of diamide or amine type ligands 62. Allylic alkylation of ester 270 with sodium dimethyl malonate 271 furnished the corresponding chiral product 272 in low yields but high enantioselectivities (up to 98%) (Scheme [68]).
4.8
Asymmetric Alkylation of β-Keto Esters
Buono and co-workers studied the use of the palladium catalyst QUIPHOS 81a as a chiral ligand in the enantioselective alkylation of β-keto esters 273 with allyl substrate 274, leading to chiral products with high enantioselectivity (up to 95% ee) depending on the nature of the substrates and specific reaction conditions. In particular, solvents such as THF led to poor enantioselectivity (5–30% ee); whereas the alkylation reaction performed with a five-membered-ring keto ester in DCM at –10 °C gave the desired product 275 in 75% yield and high enantiomeric excess (95% ee) (Scheme [69]).


4.9
Asymmetric C−H Bond Arylation Reactions
In 2019, Yu, Bertrand and co-workers studied the C–C bond coupling reaction reactivity and selectivity of quinoline-based amine ligands 278 in palladium-catalyzed β‑C(sp3)–H bond asymmetric arylation reactions. They disclosed the ligand synthesis, isolation, and detailed characterization of APAPy (acetyl-protected aminoethylpyridine) and APAQ (acetyl-protected aminoalkyl quinoline) ligands (Scheme [70]).[113]
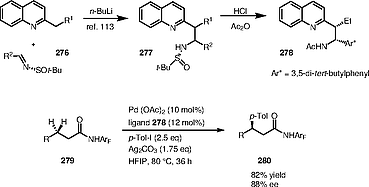
4.10
Intramolecular Aerobic Oxidative Amination of Alkenes
Stahl and co-workers (2011) described the enantioselective aerobic oxidative amination of cyclic alkenes 281 in the presence of chiral quinoline-oxazoline ligand 21d. The intramolecular addition of alkenes with a protected amine in the presence of Pd catalyst 5 mol% and chiral quinoline-oxazoline ligands 21d (7.5 mol%) gave the corresponding product in low yield but with up to 66% enantiomeric excess (Scheme [71]).

4.11
Asymmetric Oxidative Hydroboration of Alkenes
Brown and co-workers systematically studied the asymmetric hydroboration/oxidation of vinyl-arenes 284 at ambient temperature in the presence of rhodium complexes of 1,1′-(2-diarylphosphino-1-naphthyl)isoquinolines. Vinyl-arene substrates 284 bearing electron-withdrawing or -donating groups on the aryl ring led to the desired alcohol 286 with enantioselectivities up to 94% ee in the most favorable cases. The enantioselectivity of this specific conversion is moderately sensitive to the structure of the phosphorus type ligand, with the difurylphosphino ligand 121b furnishing excellent results using an electron-deficient styrene 284. Diphenylphosphino-ligand 121a showed the best results using an electron-donating alkene substrate (Scheme [72]).[75] [91]

5
Conclusions
This review compiles the advancement in the synthesis of chiral ligands containing quinoline motifs and their catalytic asymmetric reactions. The potential of chiral quinolines and their metal complexes has been demonstrated in numerous catalytic asymmetric reactions such as the addition of dialkylzinc to aldehydes and enones, addition of Grignard reagents to enones, Michael addition reactions, Friedel–Crafts alkylations, aldol lactonizations, allylic oxidations, Diels–Alder reactions, Pudovik reactions, pinacol coupling reactions, Strecker reactions, cyclopropanations of olefins, Heck reactions, hydrogenation reactions, cascade cyclizations, allylic alkylations, C–H bond arylation reactions, and oxidative hydroborations. We believe that this review will direct researchers to develop further methodologies for the synthesis of chiral quinoline-based ligands and to explore their new applications in asymmetric catalysis.
Conflict of Interest
The authors declare no conflict of interest.
-
References
- 1 Collin G, Höke H. Ullmann’s Encyclopedia of Industrial Chemistry 2012; 31: 1
- 2 Mehdi F.-M. Mini-Rev. Org. Chem. 2017; 14: 187
- 3 Bose DS, Idrees M, Jakka NM, Rao JV. J. Comb. Chem. 2010; 12: 100
- 4 Marco-Contelles J, Pérez-Mayoral E, Samadi A, Carreiras M. dC, Soriano E. Chem. Rev. 2009; 109: 2652
- 5 Bharate JB, Bharate SB, Vishwakarma RA. ACS Comb. Sci. 2014; 16: 624
- 6 Mastalir M, Glatz M, Pittenauer E, Allmaier G, Kirchner K. J. Am. Chem. Soc. 2016; 138: 15543
- 7 Beesu M, Mehta G. J. Org. Chem. 2019; 84: 8731
- 8 Willumstad TP, Boudreau PD, Danheiser RL. J. Org. Chem. 2015; 80: 11794
- 9 Weyesa A, Mulugeta E. RSC Adv. 2020; 10: 20784
- 10 Marella A, Tanwar OP, Saha R, Ali MR, Srivastava S, Akhter M, Shaquiquzzaman M, Alam MM. Saudi Pharm. J. 2013; 21: 1
- 11 Meyet CE, Larsen CH. J. Org. Chem. 2014; 79: 9835
- 12 Cretton S, Breant L, Pourrez L, Ambuehl C, Marcourt L, Ebrahimi SN, Hamburger M, Perozzo R, Karimou S, Kaiser M, Cuendet M, Christen P. J. Nat. Prod. 2014; 77: 2304
- 13 Boyd DR, Sharma ND, Loke PL, Malone JF, McRoberts WC, Hamilton JT. Org. Biomol. Chem. 2007; 5: 2983
- 14 Campbell SF, Hardstone JD, Palmer MJ. J. Med. Chem. 1988; 31: 1031
- 15 Markees DG, Dewey VC, Kidder GW. J. Med. Chem. 1970; 13: 324
- 16 Mabire D, Coupa S, Adelinet C, Poncelet A, Simonnet Y, Venet M, Wouters R, Lesage AS. J, Beijsterveldt LV, Bischoff F. J. Med. Chem. 2005; 48: 2134
- 17 Guandalini L, Norcini M, Varani K, Pistolozzi M, Gotti C, Bazzicalupi C, Martini E, Dei S, Manetti D, Scapecchi S, Teodori E, Bertucci C, Ghelardini C, Romanelli MN. J. Med. Chem. 2007; 50: 4993
- 18 Cui JJ, Shen H, Tran-Dubé M, Nambu M, McTigue M, Grodsky N, Ryan K, Yamazaki S, Aguirre S, Parker M, Li Q, Zou H, Christensen J. J. Med. Chem. 2013; 56: 6651
- 19 León B, Fong JC. N, Peach KC, Wong WR, Yildiz FH, Linington RG. Org. Lett. 2013; 15: 1234
- 20 Michael JP. Nat. Prod. Rep. 2003; 20: 476
- 21 Zhang X, Jenekhe SA. Macromolecules 2000; 33: 2069
- 22 Jenekhe SA, Lu L, Alam MM. Macromolecules 2001; 34: 7315
- 23 Zhang Z, Shi Y, Pan Y, Cheng X, Zhang L, Chen J, Li M.-J, Yi C. J. Mater. Chem. B 2014; 2: 5020
- 24 Pimpalshende DM, Dhoble SJ. Luminescence 2014; 29: 451
- 25 Biot C, Daher W, Chavain N, Fandeur T, Khalife J, Dive D, De Clercq E. J. Med. Chem. 2006; 49: 2845
- 26 Manohar S, Rajesh UC, Khan SI, Tekwani BL, Rawat DS. ACS Med. Chem. Lett. 2012; 3: 555
- 27 Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Clinic. Rev. Allerg. Immunol. 2012; 42: 145
- 28 Kumar S, Bawa S, Gupta H. Mini-Rev. Med. Chem. 2009; 9: 1648
- 29 Muruganantham N, Sivakumar R, Anbalagan N, Gunasekaran V, Leonard JT. Biol. Pharm. Bull. 2004; 27: 1683
- 30 Luchi RJ, Conn HL, Helwig J. Am. J. Cardiol. 1962; 10: 252
- 31 Nevin RL. Int. J. Parasitol. Drug. 2014; 4: 118
- 32 Dhayalan V, Gadekar SC, Alassad Z, Milo A. Nat. Chem. 2019; 11: 543
- 33 Raed AA, Dhayalan V, Barkai S, Milo A. Chimia 2020; 74: 878
- 34 Dhayalan V, Mal K, Milo A. Synthesis 2019; 51: 2845
- 35 Wang D, Weinstein AB, White PB, Stahl SS. Chem. Rev. 2018; 118: 2636
- 36 Flanigan DM, Romanov-Michailidis F, White NA, Rovis T. Chem. Rev. 2015; 115: 9307
- 37 Kurono N, Ohkuma T. ACS Catal. 2016; 6: 989
- 38 Tanriver G, Dedeoglu B, Catak S, Aviyente V. Acc. Chem. Res. 2016; 49: 1250
- 39 Chen D.-F, Han Z.-Y, Zhou X.-L, Gong L.-Z. Acc. Chem. Res. 2014; 47: 2365
- 40a Carroll M, Guiry PJ. Chem. Soc. Rev. 2014; 43: 819
- 40b Rokade BV, Barker J, Guiry PJ. Chem. Soc. Rev. 2019; 48: 4766
- 41a Rokade B, Guiry PJ. ACS Catal. 2018; 8: 624
- 41b Connon R, Roche B, Rokade BJ, Guiry PJ. Chem. Rev. 2021; 121: 6373
- 41c List B. Chem. Rev. 2007; 107: 5413
- 41d Xie Y, List B. Angew. Chem. Int. Ed. 2017; 56: 4936
- 41e Liu C, Oblak EZ, Vander Wal MN, Dilger AK, Almstead DK, MacMillan DW. C. J. Am. Chem. Soc. 2016; 138: 2134
- 41f Singh GS, Yeboah EM. O. Rep. Org. Chem. 2016; 6: 47
- 42 Shen Z.-L, Dhayalan V, Benischke AD, Greiner R, Karaghiosoff K, Mayer P, Knochel P. Angew. Chem. Int. Ed. 2016; 55: 5332
- 43 Li J, Tan E, Keller N, Chen Y.-H, Zehetmaier PM, Jakowetz AC, Bein T, Knochel P. J. Am. Chem. Soc. 2019; 141: 98
- 44 Chen Q, du Jourdin XM, Knochel P. J. Am. Chem. Soc. 2013; 135: 4958
- 45 Steib AK, Fernandez S, Kuzmina OM, Corpet M, Gosmini C, Knochel P. Synlett 2015; 26: 1049
- 46 Kuzmina OM, Steib AK, Moyeux A, Cahiez G, Knochel P. Synthesis 2015; 47: 1696
- 47 Bellan AB, Kuzmina OM, Vetsova VA, Knochel P. Synthesis 2017; 49: 188
- 48 Balkenhohl M, Valsamidou V, Knochel P. Eur. J. Org. Chem. 2019; 5165
- 49 See ref. 41a.
- 50 Cao Y, Zhang S, Antilla JC. ACS Catal. 2020; 10: 10914
- 51 Mihorianu M, Leonzio M, Monari M, Ravotto L, Ceroni P, Bettinelli M, Piccinelli F. ChemistrySelect 2016; 1: 1996
- 52 Shao Y.-D, Dong M.-M, Wang Y.-A, Cheng P.-M, Wang T, Cheng D.-J. Org. Lett. 2019; 21: 4831
- 53 Batista VF, Pinto DC. G. A, Silva AM. S. ACS Sustainable Chem. Eng. 2016; 4: 4064
- 54 Wang Q, Zhang W.-W, Song H, Wang J, Zheng C, Gu Q, You S.-L. J. Am. Chem. Soc. 2020; 142: 15678
- 55 Fernandes A, Laye C, Pramanik S, Palmeira D, Pekel Ö. Ö, Massip S, Schmidtmann M, Müller T, Robert F, Landais Y. J. Am. Chem. Soc. 2020; 142: 564
- 56 Ingalls EL, Holtzen GA, Kaminsky W, Michael FE. J. Organomet. Chem. 2017; 832: 9
- 57 Wang J, Chen MW, Ji Y, Hu SB, Zhou YG. J. Am. Chem. Soc. 2016; 138: 10413
- 58 Parvez MM, Haraguchi N, Itsuno S. Macromolecules 2014; 47: 1922
- 59 Tong M, Wang S, Zhuang J, Qin C, Li H, Wang W. Org. Lett. 2018; 20: 1195
- 60 Shao Y, Han D, Dong M.-M, Yang X, Cheng D.-J. Org. Chem. Front. 2021; 8: 605
- 61 Zheng L, Zhan Y, Yu C, Huang F, Wang Y, Jiang H. Org. Lett. 2017; 19: 1482
- 62 Wang S.-J, Wang Z, Tang Y, Chen J, Zhou L. Org. Lett. 2020; 22: 8894
- 63 Chen J, Fu Y, Yu Y, Wang J.-R, Guo Y.-W, Li H, Wang W. Org. Lett. 2020; 22: 6061
- 64 Friestad GK, Ji A, Baltrusaitis J, Korapala CS, Qin J. J. Org. Chem. 2012; 77: 3159
- 65 Thaler T, Geittner F, Knochel P. Synlett 2007; 2655
- 66 Felluga F, Baratta W, Fanfoni L, Pitacco G, Rigo P, Benedetti F. J. Org. Chem. 2009; 74: 3547
- 67 Hu X, Dawson SJ, Nagaoka Y, Tanatani A, Huc I. J. Org. Chem. 2016; 81: 1137
- 68a Kawamura K, Fukuzawa H, Hayashi M. Org. Lett. 2008; 10: 3509
- 68b Kawamura K, Fukuzawa H, Hayashi M. Bull. Chem. Soc. Jpn. 2011; 84: 640
- 69a Ramesh N, Prakash C, Sureshbabu R, Dhayalan V, Mohanakrishnan AK. Tetrahedron 2008; 64: 2071
- 69b Tan Q, Hayashi M. Adv. Synth. Catal. 2008; 350: 2639
- 69c Dhayalan V, Murakami R, Hayashi M. Asian J. Chem. 2013; 25: 7505
- 69d Hayashi M, Yamada K, Nakayama S, Hayashi H, Yamazaki S. Green Chem. 2000; 6: 257
- 69e Sano Y, Tanaka T, Hayashi M. Chem. Lett. 2007; 12: 1414
- 70a Suga H, Kakehi A, Mitsuda M. Bull. Chem. Soc. Jpn. 2004; 77: 561
- 70b Suga H, Nakajima T, Itoh K, Kakehi A. Org. Lett. 2005; 7: 1431
- 70c Shi JW, Zhao MX, Lei ZY, Shi M. J. Org. Chem. 2008; 73: 305
- 70d Suga H, Funyu A, Kakehi A. Org. Lett. 2007; 11: 97
- 71 Retmane A, Gmouh S, Runghen M, Valnot JY, Maddaluno J, Toupet L, Oulyadi H, Eddine JJ. Tetrahedron: Asymmetry 2008; 19: 1523
- 72a Chelucci G, Pinna GA, Saba A, Valenti R. Tetrahedron: Asymmetry 2000; 11: 4027
- 72b Chelucci G, Gladiali S, Saba A. Tetrahedron: Asymmetry 1999; 10: 1393
- 72c He W, Yip KT, Zhu NY, Yang D. Org. Lett. 2009; 11: 5626
- 73 Fraile JM, García JI, Osés GJ, Mayoral JA, Roldán M. Organometallics 2008; 27: 2246
- 74 Canal JM, Gómez M, Jiménez F, Rocamora M, Muller G, Duńach E, Franco D, Jiménez A, Cano FH. Organometallics 2000; 19: 966
- 75a Park SW, Son JH, Kim SG, Ahn KH. Tetrahedron: Asymmetry 1999; 10: 1903
- 75b Chelucci G, Orrù G, Pinna GA. Tetrahedron 2003; 59: 9471
- 75c Clark CR, Hay RW. J. Chem. Soc., Dalton Trans. 1974; 2148
- 76 Bolm C, Verrucci M, Simic O, Cozzi PG, Raabe G, Okamura H. Chem. Commun. 2003; 2826
- 77a Chelucci G, Saba A, Sanna G, Soccolini F. Tetrahedron: Asymmetry 2000; 11: 3427
- 77b Chelucci G, Saba A. Tetrahedron: Asymmetry 1998; 9: 2575
- 78a Takenaka N, Xia G, Yamamoto H. J. Am. Chem. Soc. 2004; 126: 13198
- 78b Abell JP, Yamamoto H. J. Am. Chem. Soc. 2008; 130: 10521
- 79 See ref. 17.
- 80 Kwong HL, Yeung HL, Yeung CT, Lee WS, Lee CS, Wong WL. Coord. Chem. Rev. 2007; 251: 2188
- 81a Qi G, Judeh ZM. A. Tetrahedron: Asymmetry 2010; 21: 429
- 81b Qi G, Ji YQ, Judeh ZM. A. Tetrahedron 2010; 66: 4195
- 82 Martinez R, Zoli L, Cozzi PG, Ramon DJ, Yus M. Tetrahedron: Asymmetry 2008; 19: 2600
- 83a Baratta W, Fanfoni L, Magnolia S, Siega K, Rigo P. Eur. J. Inorg. Chem. 2010; 1419
- 83b Baratta W, Ballico M, Baldino S, Chelucci G, Herdtweck E, Siega K, Magnolia S, Rigo P. Chem. Eur. J. 2008; 14: 9148
- 83c See ref. 66.
- 84a Delapierre G, Brunel JM, Constantieux T, Buono G. Tetrahedron: Asymmetry 2001; 12: 1345
- 84b Delapierre G, Constantieux T, Brunel JM, Buono G. Eur. J. Org. Chem. 2000; 2507
- 84c Brunel JM, Constantieux T, Buono G. J. Org. Chem. 1999; 64: 8940
- 85 Delapierre G, Achard M, Buono G. Tetrahedron Lett. 2002; 43: 4025
- 86 Franciò G, Drommi D, Graiff C, Faraone F, Tiripicchio A. Inorg. Chim. Acta 2002; 338: 59
- 87a Franciò G, Arena CG, Faraone F, Graiff C, Lanfranchi M, Tiripicchio A. Eur. J. Inorg. Chem. 1999; 1219
- 87b Franciò G, Faraone F, Leitner W. Angew. Chem. Int. Ed. 2000; 39: 1428
- 88a Dhayalan V, Sämann C, Knochel P. Chem. Commun. 2015; 51: 3239
- 88b Dhayalan V, Alcañiz FR, Werner V, Karaghiosoff K, Knochel P. Synthesis 2015; 47: 3972
- 88c Sämann C, Dhayalan V, Schreiner PR, Knochel P. Org. Lett. 2014; 16: 2418
- 88d Schlücker T, Dhayalan V, Langhals H, Sämann C, Knochel P. Asian J. Org. Chem. 2015; 4: 763
- 88e Bunlaksananusorn T, Knochel P. J. Org. Chem. 2004; 69: 4595
- 88f See ref. 65.
- 89 Jiang B, Lei Y, Zhao XL. J. Org. Chem. 2008; 73: 7833
- 90 Ruzziconi R, Santi C, Spizzichino S. Tetrahedron: Asymmetry 2007; 18: 1742
- 91a Alcock NW, Brown JM, Htimes DI. Tetrahedron: Asymmetry 1993; 4: 743
- 91b Valk JM, Whitlock GA, Layzell TP, Brown JM. Tetrahedron: Asymmetry 1995; 6: 2593
- 91c Doucet H, Fernandez E, Layzell TP, Brown JM. Chem. Eur. J. 1999; 5: 1320
- 91d See ref. 75b.
- 92a Markò IE, Vanherck JC, Ates A, Tinant B, Declercq JP. Tetrahedron Lett. 2003; 44: 3333
- 92b Han Z, Wang Z, Zhang X, Ding K. Tetrahedron: Asymmetry 2010; 21: 1529
- 93 Drury WJ. III, Zimmermann N, Keenan M, Hayashi M, Kaiser S, Goddard R, Pfaltz A. Angew. Chem. Int. Ed. 2004; 43: 70
- 94a Chelucci G, Orrù G. Tetrahedron Lett. 2005; 46: 3493
- 94b Chelucci G, Baldino S. Tetrahedron: Asymmetry 2006; 17: 1529
- 95 Allen DG, Mclaughlin GM, Robertson GB, Steffen WL, Salem G, Wild SB. Inorg. Chem. 1982; 21: 1007
- 96a Thummel RP, Lefoulon F. J. Org. Chem. 1985; 50: 666
- 96b Chelucci G, Thummel RP. Chem. Rev. 2002; 102: 3129
- 96c Saito M, Nakajima M, Hashimoto S. Chem. Commun. 2000; 1851
- 96d Saito M, Nakajima M, Hashimoto S. Tetrahedron 2000; 56: 9589
- 96e Chelucci G, Murineddu G, Pinna GA. Tetrahedron: Asymmetry 2004; 15: 1373
- 96f Malkov AV, Dufková L, Farrugia L, Koćovský P. Angew. Chem. Int. Ed. 2003; 42: 3674
- 96g Jiao Z, Feng X, Liu B, Chen F, Zhang G, Jiang Y. Eur. J. Org. Chem. 2003; 3818
- 96h Liu B, Feng X, Chen F, Zhang G, Cui X, Jiang Y. Synlett 2001; 1551
- 97a Dhayalan V, Knochel P. Synthesis 2015; 47: 3246
- 97b Meyers AI, Wettlaufer DG. J. Am. Chem. Soc. 1984; 106: 1135
- 97c Meyers AI. J. Org. Chem. 2005; 70: 6137
- 98 Arena CG, Calabro G, Francio G, Faraone F. Tetrahedron: Asymmetry 2000; 11: 2387
- 99 Qi G, Judeh ZM. A. Synth. Commun. 2012; 42: 1585
- 100 Nakajima M, Yamamoto S, Yamaguchi Y, Nakamura S, Hashimoto S. Tetrahedron 2003; 59: 7307
- 101 Jia YX, Zhu SF, Yang Y, Zhou QL. J. Org. Chem. 2006; 71: 75
- 102a Tan Q, Hayashi M. Org. Lett. 2009; 11: 3314
- 102b Tanaka T, Tan Q, Iwanaga K, Hayashi M. Carbohydr. Res. 2011; 346: 340
- 103a Malkov AV, Orsini M, Pernazza D, Muir KW, Langer V, Meghani P, Koćovský P. Org. Lett. 2002; 4: 1047
- 103b Wrzeszcz Z, Siedlecka R. Catalysts 2021; 4: 444
- 104 Brunel JM, Campo BD, Buono G. Tetrahedron Lett. 1998; 39: 9663
- 105 Suga H, Furihata Y, Sakamoto A, Itoh K, Yukihisa OkumuraY, Tsuchida T, Kakehi A, Baba T. J. Org. Chem. 2011; 76: 7377
- 106 Fraile JM, García JI, José A, Mayoral JA, Roldán M. Org. Lett. 2007; 9: 731
- 107 See ref. 73.
- 108 Burk S, Franciò G, Leitner W. Chem. Commun. 2005; 3460
- 109a Zhang Y, Sigman MS. J. Am. Chem. Soc. 2007; 129: 3076
- 109b Jensen KH, Webb JD, Sigman MS. J. Am. Chem. Soc. 2010; 132: 17471
- 110a Zhang R, Xie B, Chen G.-S, Qiu L, Chen Y.-X. Tetrahedron Lett. 2016; 57: 845
- 110b Pamìes O, Margalef J, Cañellas S, James J, Judge J, Guiry PJ, Moberg C, Bäckvall J.-E, Pfaltz A, Pericas MA, Diéguez M. Chem. Rev. 2021; 121: 4373
- 110c Chelucci G, Medici S, Saba A. Tetrahedron: Asymmetry 1999; 10: 543
- 110d Fekner T, Bunz HM, Guiry PJ. Eur. J. Org. Chem. 2008; 5055
- 110e Drommi D, Saporita M, Bruno G, Faraone F, Scafato P, Rosini C. Dalton Trans. 2007; 1509
- 110f Sureshbabu R, Saravanan V, Dhayalan V, Mohanakrishnan AK. Eur. J. Org. Chem. 2011; 922
- 110g Dhayalan V, Clement JA, Jagan R, Mohanakrishnan AK. Eur. J. Org. Chem. 2009; 531
- 110h Mohanakrishnan AK, Dhayalan V, Clement JA, Sureshbabu RB. R, Kumar NS. Tetrahedron Lett. 2008; 49: 5850
- 111 Trost BM, Dogra K, Hachiya I, Emura T, Hughes DL, Krska S, Reamer RA, Palucki M, Yasuda N, Reider PJ. Angew. Chem. Int. Ed. 2002; 41: 1929
- 112 Brunel JM, Tenaglia A, Buono G. Tetrahedron: Asymmetry 2000; 11: 3585
- 113a Romero EA, Chen G, Gembicky M, Jazzar R, Yu J.-Q, Bertrand G. J. Am. Chem. Soc. 2019; 141: 16726
- 113b Andrä MS, Schifferer L, Pollok CH, Merten C, Gooßen LJ, Yu J.-Q. Chem. Eur. J. 2019; 25: 8503
- 114 McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org. Lett. 2011; 13: 2830
Corresponding Author
Publication History
Received: 15 November 2021
Accepted after revision: 12 January 2022
Accepted Manuscript online:
18 January 2022
Article published online:
08 February 2022
© 2022. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/)
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
References
- 1 Collin G, Höke H. Ullmann’s Encyclopedia of Industrial Chemistry 2012; 31: 1
- 2 Mehdi F.-M. Mini-Rev. Org. Chem. 2017; 14: 187
- 3 Bose DS, Idrees M, Jakka NM, Rao JV. J. Comb. Chem. 2010; 12: 100
- 4 Marco-Contelles J, Pérez-Mayoral E, Samadi A, Carreiras M. dC, Soriano E. Chem. Rev. 2009; 109: 2652
- 5 Bharate JB, Bharate SB, Vishwakarma RA. ACS Comb. Sci. 2014; 16: 624
- 6 Mastalir M, Glatz M, Pittenauer E, Allmaier G, Kirchner K. J. Am. Chem. Soc. 2016; 138: 15543
- 7 Beesu M, Mehta G. J. Org. Chem. 2019; 84: 8731
- 8 Willumstad TP, Boudreau PD, Danheiser RL. J. Org. Chem. 2015; 80: 11794
- 9 Weyesa A, Mulugeta E. RSC Adv. 2020; 10: 20784
- 10 Marella A, Tanwar OP, Saha R, Ali MR, Srivastava S, Akhter M, Shaquiquzzaman M, Alam MM. Saudi Pharm. J. 2013; 21: 1
- 11 Meyet CE, Larsen CH. J. Org. Chem. 2014; 79: 9835
- 12 Cretton S, Breant L, Pourrez L, Ambuehl C, Marcourt L, Ebrahimi SN, Hamburger M, Perozzo R, Karimou S, Kaiser M, Cuendet M, Christen P. J. Nat. Prod. 2014; 77: 2304
- 13 Boyd DR, Sharma ND, Loke PL, Malone JF, McRoberts WC, Hamilton JT. Org. Biomol. Chem. 2007; 5: 2983
- 14 Campbell SF, Hardstone JD, Palmer MJ. J. Med. Chem. 1988; 31: 1031
- 15 Markees DG, Dewey VC, Kidder GW. J. Med. Chem. 1970; 13: 324
- 16 Mabire D, Coupa S, Adelinet C, Poncelet A, Simonnet Y, Venet M, Wouters R, Lesage AS. J, Beijsterveldt LV, Bischoff F. J. Med. Chem. 2005; 48: 2134
- 17 Guandalini L, Norcini M, Varani K, Pistolozzi M, Gotti C, Bazzicalupi C, Martini E, Dei S, Manetti D, Scapecchi S, Teodori E, Bertucci C, Ghelardini C, Romanelli MN. J. Med. Chem. 2007; 50: 4993
- 18 Cui JJ, Shen H, Tran-Dubé M, Nambu M, McTigue M, Grodsky N, Ryan K, Yamazaki S, Aguirre S, Parker M, Li Q, Zou H, Christensen J. J. Med. Chem. 2013; 56: 6651
- 19 León B, Fong JC. N, Peach KC, Wong WR, Yildiz FH, Linington RG. Org. Lett. 2013; 15: 1234
- 20 Michael JP. Nat. Prod. Rep. 2003; 20: 476
- 21 Zhang X, Jenekhe SA. Macromolecules 2000; 33: 2069
- 22 Jenekhe SA, Lu L, Alam MM. Macromolecules 2001; 34: 7315
- 23 Zhang Z, Shi Y, Pan Y, Cheng X, Zhang L, Chen J, Li M.-J, Yi C. J. Mater. Chem. B 2014; 2: 5020
- 24 Pimpalshende DM, Dhoble SJ. Luminescence 2014; 29: 451
- 25 Biot C, Daher W, Chavain N, Fandeur T, Khalife J, Dive D, De Clercq E. J. Med. Chem. 2006; 49: 2845
- 26 Manohar S, Rajesh UC, Khan SI, Tekwani BL, Rawat DS. ACS Med. Chem. Lett. 2012; 3: 555
- 27 Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Clinic. Rev. Allerg. Immunol. 2012; 42: 145
- 28 Kumar S, Bawa S, Gupta H. Mini-Rev. Med. Chem. 2009; 9: 1648
- 29 Muruganantham N, Sivakumar R, Anbalagan N, Gunasekaran V, Leonard JT. Biol. Pharm. Bull. 2004; 27: 1683
- 30 Luchi RJ, Conn HL, Helwig J. Am. J. Cardiol. 1962; 10: 252
- 31 Nevin RL. Int. J. Parasitol. Drug. 2014; 4: 118
- 32 Dhayalan V, Gadekar SC, Alassad Z, Milo A. Nat. Chem. 2019; 11: 543
- 33 Raed AA, Dhayalan V, Barkai S, Milo A. Chimia 2020; 74: 878
- 34 Dhayalan V, Mal K, Milo A. Synthesis 2019; 51: 2845
- 35 Wang D, Weinstein AB, White PB, Stahl SS. Chem. Rev. 2018; 118: 2636
- 36 Flanigan DM, Romanov-Michailidis F, White NA, Rovis T. Chem. Rev. 2015; 115: 9307
- 37 Kurono N, Ohkuma T. ACS Catal. 2016; 6: 989
- 38 Tanriver G, Dedeoglu B, Catak S, Aviyente V. Acc. Chem. Res. 2016; 49: 1250
- 39 Chen D.-F, Han Z.-Y, Zhou X.-L, Gong L.-Z. Acc. Chem. Res. 2014; 47: 2365
- 40a Carroll M, Guiry PJ. Chem. Soc. Rev. 2014; 43: 819
- 40b Rokade BV, Barker J, Guiry PJ. Chem. Soc. Rev. 2019; 48: 4766
- 41a Rokade B, Guiry PJ. ACS Catal. 2018; 8: 624
- 41b Connon R, Roche B, Rokade BJ, Guiry PJ. Chem. Rev. 2021; 121: 6373
- 41c List B. Chem. Rev. 2007; 107: 5413
- 41d Xie Y, List B. Angew. Chem. Int. Ed. 2017; 56: 4936
- 41e Liu C, Oblak EZ, Vander Wal MN, Dilger AK, Almstead DK, MacMillan DW. C. J. Am. Chem. Soc. 2016; 138: 2134
- 41f Singh GS, Yeboah EM. O. Rep. Org. Chem. 2016; 6: 47
- 42 Shen Z.-L, Dhayalan V, Benischke AD, Greiner R, Karaghiosoff K, Mayer P, Knochel P. Angew. Chem. Int. Ed. 2016; 55: 5332
- 43 Li J, Tan E, Keller N, Chen Y.-H, Zehetmaier PM, Jakowetz AC, Bein T, Knochel P. J. Am. Chem. Soc. 2019; 141: 98
- 44 Chen Q, du Jourdin XM, Knochel P. J. Am. Chem. Soc. 2013; 135: 4958
- 45 Steib AK, Fernandez S, Kuzmina OM, Corpet M, Gosmini C, Knochel P. Synlett 2015; 26: 1049
- 46 Kuzmina OM, Steib AK, Moyeux A, Cahiez G, Knochel P. Synthesis 2015; 47: 1696
- 47 Bellan AB, Kuzmina OM, Vetsova VA, Knochel P. Synthesis 2017; 49: 188
- 48 Balkenhohl M, Valsamidou V, Knochel P. Eur. J. Org. Chem. 2019; 5165
- 49 See ref. 41a.
- 50 Cao Y, Zhang S, Antilla JC. ACS Catal. 2020; 10: 10914
- 51 Mihorianu M, Leonzio M, Monari M, Ravotto L, Ceroni P, Bettinelli M, Piccinelli F. ChemistrySelect 2016; 1: 1996
- 52 Shao Y.-D, Dong M.-M, Wang Y.-A, Cheng P.-M, Wang T, Cheng D.-J. Org. Lett. 2019; 21: 4831
- 53 Batista VF, Pinto DC. G. A, Silva AM. S. ACS Sustainable Chem. Eng. 2016; 4: 4064
- 54 Wang Q, Zhang W.-W, Song H, Wang J, Zheng C, Gu Q, You S.-L. J. Am. Chem. Soc. 2020; 142: 15678
- 55 Fernandes A, Laye C, Pramanik S, Palmeira D, Pekel Ö. Ö, Massip S, Schmidtmann M, Müller T, Robert F, Landais Y. J. Am. Chem. Soc. 2020; 142: 564
- 56 Ingalls EL, Holtzen GA, Kaminsky W, Michael FE. J. Organomet. Chem. 2017; 832: 9
- 57 Wang J, Chen MW, Ji Y, Hu SB, Zhou YG. J. Am. Chem. Soc. 2016; 138: 10413
- 58 Parvez MM, Haraguchi N, Itsuno S. Macromolecules 2014; 47: 1922
- 59 Tong M, Wang S, Zhuang J, Qin C, Li H, Wang W. Org. Lett. 2018; 20: 1195
- 60 Shao Y, Han D, Dong M.-M, Yang X, Cheng D.-J. Org. Chem. Front. 2021; 8: 605
- 61 Zheng L, Zhan Y, Yu C, Huang F, Wang Y, Jiang H. Org. Lett. 2017; 19: 1482
- 62 Wang S.-J, Wang Z, Tang Y, Chen J, Zhou L. Org. Lett. 2020; 22: 8894
- 63 Chen J, Fu Y, Yu Y, Wang J.-R, Guo Y.-W, Li H, Wang W. Org. Lett. 2020; 22: 6061
- 64 Friestad GK, Ji A, Baltrusaitis J, Korapala CS, Qin J. J. Org. Chem. 2012; 77: 3159
- 65 Thaler T, Geittner F, Knochel P. Synlett 2007; 2655
- 66 Felluga F, Baratta W, Fanfoni L, Pitacco G, Rigo P, Benedetti F. J. Org. Chem. 2009; 74: 3547
- 67 Hu X, Dawson SJ, Nagaoka Y, Tanatani A, Huc I. J. Org. Chem. 2016; 81: 1137
- 68a Kawamura K, Fukuzawa H, Hayashi M. Org. Lett. 2008; 10: 3509
- 68b Kawamura K, Fukuzawa H, Hayashi M. Bull. Chem. Soc. Jpn. 2011; 84: 640
- 69a Ramesh N, Prakash C, Sureshbabu R, Dhayalan V, Mohanakrishnan AK. Tetrahedron 2008; 64: 2071
- 69b Tan Q, Hayashi M. Adv. Synth. Catal. 2008; 350: 2639
- 69c Dhayalan V, Murakami R, Hayashi M. Asian J. Chem. 2013; 25: 7505
- 69d Hayashi M, Yamada K, Nakayama S, Hayashi H, Yamazaki S. Green Chem. 2000; 6: 257
- 69e Sano Y, Tanaka T, Hayashi M. Chem. Lett. 2007; 12: 1414
- 70a Suga H, Kakehi A, Mitsuda M. Bull. Chem. Soc. Jpn. 2004; 77: 561
- 70b Suga H, Nakajima T, Itoh K, Kakehi A. Org. Lett. 2005; 7: 1431
- 70c Shi JW, Zhao MX, Lei ZY, Shi M. J. Org. Chem. 2008; 73: 305
- 70d Suga H, Funyu A, Kakehi A. Org. Lett. 2007; 11: 97
- 71 Retmane A, Gmouh S, Runghen M, Valnot JY, Maddaluno J, Toupet L, Oulyadi H, Eddine JJ. Tetrahedron: Asymmetry 2008; 19: 1523
- 72a Chelucci G, Pinna GA, Saba A, Valenti R. Tetrahedron: Asymmetry 2000; 11: 4027
- 72b Chelucci G, Gladiali S, Saba A. Tetrahedron: Asymmetry 1999; 10: 1393
- 72c He W, Yip KT, Zhu NY, Yang D. Org. Lett. 2009; 11: 5626
- 73 Fraile JM, García JI, Osés GJ, Mayoral JA, Roldán M. Organometallics 2008; 27: 2246
- 74 Canal JM, Gómez M, Jiménez F, Rocamora M, Muller G, Duńach E, Franco D, Jiménez A, Cano FH. Organometallics 2000; 19: 966
- 75a Park SW, Son JH, Kim SG, Ahn KH. Tetrahedron: Asymmetry 1999; 10: 1903
- 75b Chelucci G, Orrù G, Pinna GA. Tetrahedron 2003; 59: 9471
- 75c Clark CR, Hay RW. J. Chem. Soc., Dalton Trans. 1974; 2148
- 76 Bolm C, Verrucci M, Simic O, Cozzi PG, Raabe G, Okamura H. Chem. Commun. 2003; 2826
- 77a Chelucci G, Saba A, Sanna G, Soccolini F. Tetrahedron: Asymmetry 2000; 11: 3427
- 77b Chelucci G, Saba A. Tetrahedron: Asymmetry 1998; 9: 2575
- 78a Takenaka N, Xia G, Yamamoto H. J. Am. Chem. Soc. 2004; 126: 13198
- 78b Abell JP, Yamamoto H. J. Am. Chem. Soc. 2008; 130: 10521
- 79 See ref. 17.
- 80 Kwong HL, Yeung HL, Yeung CT, Lee WS, Lee CS, Wong WL. Coord. Chem. Rev. 2007; 251: 2188
- 81a Qi G, Judeh ZM. A. Tetrahedron: Asymmetry 2010; 21: 429
- 81b Qi G, Ji YQ, Judeh ZM. A. Tetrahedron 2010; 66: 4195
- 82 Martinez R, Zoli L, Cozzi PG, Ramon DJ, Yus M. Tetrahedron: Asymmetry 2008; 19: 2600
- 83a Baratta W, Fanfoni L, Magnolia S, Siega K, Rigo P. Eur. J. Inorg. Chem. 2010; 1419
- 83b Baratta W, Ballico M, Baldino S, Chelucci G, Herdtweck E, Siega K, Magnolia S, Rigo P. Chem. Eur. J. 2008; 14: 9148
- 83c See ref. 66.
- 84a Delapierre G, Brunel JM, Constantieux T, Buono G. Tetrahedron: Asymmetry 2001; 12: 1345
- 84b Delapierre G, Constantieux T, Brunel JM, Buono G. Eur. J. Org. Chem. 2000; 2507
- 84c Brunel JM, Constantieux T, Buono G. J. Org. Chem. 1999; 64: 8940
- 85 Delapierre G, Achard M, Buono G. Tetrahedron Lett. 2002; 43: 4025
- 86 Franciò G, Drommi D, Graiff C, Faraone F, Tiripicchio A. Inorg. Chim. Acta 2002; 338: 59
- 87a Franciò G, Arena CG, Faraone F, Graiff C, Lanfranchi M, Tiripicchio A. Eur. J. Inorg. Chem. 1999; 1219
- 87b Franciò G, Faraone F, Leitner W. Angew. Chem. Int. Ed. 2000; 39: 1428
- 88a Dhayalan V, Sämann C, Knochel P. Chem. Commun. 2015; 51: 3239
- 88b Dhayalan V, Alcañiz FR, Werner V, Karaghiosoff K, Knochel P. Synthesis 2015; 47: 3972
- 88c Sämann C, Dhayalan V, Schreiner PR, Knochel P. Org. Lett. 2014; 16: 2418
- 88d Schlücker T, Dhayalan V, Langhals H, Sämann C, Knochel P. Asian J. Org. Chem. 2015; 4: 763
- 88e Bunlaksananusorn T, Knochel P. J. Org. Chem. 2004; 69: 4595
- 88f See ref. 65.
- 89 Jiang B, Lei Y, Zhao XL. J. Org. Chem. 2008; 73: 7833
- 90 Ruzziconi R, Santi C, Spizzichino S. Tetrahedron: Asymmetry 2007; 18: 1742
- 91a Alcock NW, Brown JM, Htimes DI. Tetrahedron: Asymmetry 1993; 4: 743
- 91b Valk JM, Whitlock GA, Layzell TP, Brown JM. Tetrahedron: Asymmetry 1995; 6: 2593
- 91c Doucet H, Fernandez E, Layzell TP, Brown JM. Chem. Eur. J. 1999; 5: 1320
- 91d See ref. 75b.
- 92a Markò IE, Vanherck JC, Ates A, Tinant B, Declercq JP. Tetrahedron Lett. 2003; 44: 3333
- 92b Han Z, Wang Z, Zhang X, Ding K. Tetrahedron: Asymmetry 2010; 21: 1529
- 93 Drury WJ. III, Zimmermann N, Keenan M, Hayashi M, Kaiser S, Goddard R, Pfaltz A. Angew. Chem. Int. Ed. 2004; 43: 70
- 94a Chelucci G, Orrù G. Tetrahedron Lett. 2005; 46: 3493
- 94b Chelucci G, Baldino S. Tetrahedron: Asymmetry 2006; 17: 1529
- 95 Allen DG, Mclaughlin GM, Robertson GB, Steffen WL, Salem G, Wild SB. Inorg. Chem. 1982; 21: 1007
- 96a Thummel RP, Lefoulon F. J. Org. Chem. 1985; 50: 666
- 96b Chelucci G, Thummel RP. Chem. Rev. 2002; 102: 3129
- 96c Saito M, Nakajima M, Hashimoto S. Chem. Commun. 2000; 1851
- 96d Saito M, Nakajima M, Hashimoto S. Tetrahedron 2000; 56: 9589
- 96e Chelucci G, Murineddu G, Pinna GA. Tetrahedron: Asymmetry 2004; 15: 1373
- 96f Malkov AV, Dufková L, Farrugia L, Koćovský P. Angew. Chem. Int. Ed. 2003; 42: 3674
- 96g Jiao Z, Feng X, Liu B, Chen F, Zhang G, Jiang Y. Eur. J. Org. Chem. 2003; 3818
- 96h Liu B, Feng X, Chen F, Zhang G, Cui X, Jiang Y. Synlett 2001; 1551
- 97a Dhayalan V, Knochel P. Synthesis 2015; 47: 3246
- 97b Meyers AI, Wettlaufer DG. J. Am. Chem. Soc. 1984; 106: 1135
- 97c Meyers AI. J. Org. Chem. 2005; 70: 6137
- 98 Arena CG, Calabro G, Francio G, Faraone F. Tetrahedron: Asymmetry 2000; 11: 2387
- 99 Qi G, Judeh ZM. A. Synth. Commun. 2012; 42: 1585
- 100 Nakajima M, Yamamoto S, Yamaguchi Y, Nakamura S, Hashimoto S. Tetrahedron 2003; 59: 7307
- 101 Jia YX, Zhu SF, Yang Y, Zhou QL. J. Org. Chem. 2006; 71: 75
- 102a Tan Q, Hayashi M. Org. Lett. 2009; 11: 3314
- 102b Tanaka T, Tan Q, Iwanaga K, Hayashi M. Carbohydr. Res. 2011; 346: 340
- 103a Malkov AV, Orsini M, Pernazza D, Muir KW, Langer V, Meghani P, Koćovský P. Org. Lett. 2002; 4: 1047
- 103b Wrzeszcz Z, Siedlecka R. Catalysts 2021; 4: 444
- 104 Brunel JM, Campo BD, Buono G. Tetrahedron Lett. 1998; 39: 9663
- 105 Suga H, Furihata Y, Sakamoto A, Itoh K, Yukihisa OkumuraY, Tsuchida T, Kakehi A, Baba T. J. Org. Chem. 2011; 76: 7377
- 106 Fraile JM, García JI, José A, Mayoral JA, Roldán M. Org. Lett. 2007; 9: 731
- 107 See ref. 73.
- 108 Burk S, Franciò G, Leitner W. Chem. Commun. 2005; 3460
- 109a Zhang Y, Sigman MS. J. Am. Chem. Soc. 2007; 129: 3076
- 109b Jensen KH, Webb JD, Sigman MS. J. Am. Chem. Soc. 2010; 132: 17471
- 110a Zhang R, Xie B, Chen G.-S, Qiu L, Chen Y.-X. Tetrahedron Lett. 2016; 57: 845
- 110b Pamìes O, Margalef J, Cañellas S, James J, Judge J, Guiry PJ, Moberg C, Bäckvall J.-E, Pfaltz A, Pericas MA, Diéguez M. Chem. Rev. 2021; 121: 4373
- 110c Chelucci G, Medici S, Saba A. Tetrahedron: Asymmetry 1999; 10: 543
- 110d Fekner T, Bunz HM, Guiry PJ. Eur. J. Org. Chem. 2008; 5055
- 110e Drommi D, Saporita M, Bruno G, Faraone F, Scafato P, Rosini C. Dalton Trans. 2007; 1509
- 110f Sureshbabu R, Saravanan V, Dhayalan V, Mohanakrishnan AK. Eur. J. Org. Chem. 2011; 922
- 110g Dhayalan V, Clement JA, Jagan R, Mohanakrishnan AK. Eur. J. Org. Chem. 2009; 531
- 110h Mohanakrishnan AK, Dhayalan V, Clement JA, Sureshbabu RB. R, Kumar NS. Tetrahedron Lett. 2008; 49: 5850
- 111 Trost BM, Dogra K, Hachiya I, Emura T, Hughes DL, Krska S, Reamer RA, Palucki M, Yasuda N, Reider PJ. Angew. Chem. Int. Ed. 2002; 41: 1929
- 112 Brunel JM, Tenaglia A, Buono G. Tetrahedron: Asymmetry 2000; 11: 3585
- 113a Romero EA, Chen G, Gembicky M, Jazzar R, Yu J.-Q, Bertrand G. J. Am. Chem. Soc. 2019; 141: 16726
- 113b Andrä MS, Schifferer L, Pollok CH, Merten C, Gooßen LJ, Yu J.-Q. Chem. Eur. J. 2019; 25: 8503
- 114 McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org. Lett. 2011; 13: 2830