Subscribe to RSS
DOI: 10.1055/a-1384-4641
Seltene Erkrankungen der Orbita
Article in several languages: deutsch | English
- Zusammenfassung
- 1. Allgemeine Vorbetrachtungen
- 2. Interdisziplinarität und „Was ist selten, was ist häufig?“ – ein Dilemma?
- 3. Anamnese und Diagnostik
- 4. Strukturelle Läsionen der Orbita
- 4.4 Knöcherne Anomalien und mesodermale Defekte
- 5. Entzündliche Läsionen der Orbita
- 5.1 Infektiöse orbitale Entzündungen
- 5.2 Nicht infektiöse orbitale Entzündungen
- 5.2.3 Vaskulitiden im Bereich der Orbita
- 6. Degenerative Erkrankungen
- 7. Tumoren der Orbita 2 201 233
- 7.2 Spezieller Teil
- 7.2.1 Neoplasien der Tränendrüse
- Literatur
Zusammenfassung
Dieser Übersichtsartikel stellt eine Zusammenstellung seltener Erkrankungen der Orbita dar. Es werden angeborene Fehlbildungen und strukturelle Läsionen, entzündliche Erkrankungen, gutartige und bösartige epitheliale sowie nicht epitheliale Tumoren erörtert. Da in der Orbita zahlreiche Gewebetypen auf engstem Raum beieinanderliegen, kommen dort besonders viele Erkrankungen vor, sodass der Vollständigkeit und dem Tiefgang bei den einzelnen Abhandlungen natürliche Grenzen gesetzt sind. Die Kapitel beinhalten eine Übersicht über das jeweilige klinische Erscheinungsbild, wichtige Stichpunkte zur Diagnostik, Therapie und gegebenenfalls zur Prognose der Erkrankung. Dabei wurde versucht, die besonders charakteristischen Merkmale der einzelnen Erkrankungen herauszuarbeiten, sodass trotz der kurzen Darstellung die relevanten Aspekte enthalten sind.
1. Allgemeine Vorbetrachtungen
Erkrankungen der Orbita begegnen uns regelmäßig im klinischen Alltag. Die komplexe Anatomie mit der Vielzahl an verschiedenen Geweben führt zu einer immensen Heterogenität an Pathologien. Zahlreiche Systemerkrankungen weisen Assoziationen zu dieser Region auf. Hinzu kommen verschiedene Fehlbildungen, welche einer komplexen Embryologie entspringen. Insofern ist die Betreuung von Patienten mit Erkrankungen der Orbita als ein Paradebeispiel für interdisziplinäres Zusammenarbeiten anzusehen. Die zentrale Lage im Gesichtsschädel und die enge topografische Beziehung zur Nase mit den Nasennebenhöhlen, zur Schädelbasis und dem Hirnschädel sowie zur Schläfenregion erfordert zudem oftmals auch eine fächerübergreifende chirurgische Betreuung.
Die Orbita dient dem Schutz des Auges und seiner Anhangsgebilde. Die recht stabile knöcherne Umrahmung mit dem darin befindlichen Corpus adiposum orbitae bietet dem Organ nicht nur Sicherheit. Durch den vorgegebenen Raum können Volumenexpansionen schnell zu einem Platzproblem führen. Diese Konstellation bringt es mit sich, dass die meisten Krankheitsprozesse der Orbita sich primär ophthalmologisch manifestieren. Dabei sind Exophthalmus, Diplopie und Visusabfall als häufigste klinische Zeichen zu nennen. Dem Augenarzt kommt somit eine wichtige Erkennungs- und Steuerfunktion zu.
Prinzipiell sind uns viele Erkrankungen der Orbita wohl bekannt, beispielsweise die endokrine Orbitopathie. Dabei sollte nicht unerwähnt bleiben, dass auch diese Erkrankungen untypische und seltene Verläufe nehmen können. Demgegenüber gibt es wirkliche Raritäten. Die Vielfalt an seltenen Erkrankungen ist sehr groß und es ist nicht Ziel dieses Referates, an dieser Stelle Anspruch auf Vollständigkeit zu erheben. Die meisten der hier genannten Erkrankungen werden dem Kliniker womöglich niemals begegnen. Dennoch erscheint es den Autoren sinnvoll, die Mannigfaltigkeit und Variabilität der Erkrankungen dieser kleinen Körperregion möglichst umfassend darzustellen. Hierdurch soll der Beitrag den klinischen Blick schärfen und bei der erstmaligen Konfrontation mit womöglich bisher unbekannten Manifestationen eine Hilfestellung geben können.
Weiterhin soll in diesem Referat nicht auf periorbitale (präseptale) Erkrankungen eingegangen werden. Demnach werden Pathologien der Lider und der ableitenden Tränenwege ausgespart. Auch bleibt die Traumatologie der Orbita unbeleuchtet. Des Weiteren wird bewusst nicht auf rein okuläre Erkrankungen eingegangen, wohl wissend, dass diese über die okulären Grenzstrukturen (z. B. die Sklera) hinaus expandieren oder eine orbitale Begleitreaktion hervorrufen können.
2. Interdisziplinarität und „Was ist selten, was ist häufig?“ – ein Dilemma?
Die Perspektive auf die Orbita und die Wahrnehmung von Häufigkeiten ist fachspezifisch sehr unterschiedlich. Dies spiegelt sich auch in der Literatur wider. Orbitaprozesse kommen insgesamt nicht sehr oft vor, sodass es schwierig ist, genaue Häufigkeiten bei unterschiedlichen Patientenkollektiven der mitbehandelnden Fachdisziplinen zu benennen. Auch gibt es keine größeren randomisierten Studien. Zumeist handelt es sich um vergleichende Studien mit kleinen Fallzahlen. Bisweilen liegen nur nicht vergleichende Untersuchungen oder wenige Fallberichte vor.
Wir haben mit der Orbita ein kleines begrenztes Gebiet des menschlichen Körpers, welches jedoch zahlreiche Gewebe beherbergt und von den Fachdisziplinen der Kopf-Hals-Region betreut wird. Chirurgisch spielen hier die Kollegen der Neurochirurgie, der Mund-Kiefer-Gesichtschirurgie, der HNO und der Augenheilkunde die wesentlichen Rollen. Nicht unerwähnt sollen aber auch die zahlreichen verschiedenen Fachdisziplinen bleiben, ohne die ein gutes Patientenmanagement undenkbar wäre, u. a. die Innere Medizin, die Pädiatrie und die radiologischen und hämatoonokologischen Fächer. Zuletzt sei auf die große Bedeutung der Pathologie hingewiesen, welche durch moderne Methoden (z. B. Molekularpathologie) einen unerlässlichen Anteil zur Diagnosefindung beiträgt.
Die Orbitachirurgie stellt ein Grenzgebiet verschiedener Fachrichtungen entsprechend der anatomisch angrenzenden Strukturen dar. Es handelt sich um eine interdisziplinär zu behandelnde Körperregion.
3. Anamnese und Diagnostik
3.1 Anamnese
Der gezielten Diagnostik und Therapie im Bereich der Orbita geht eine strukturierte Anamnese voraus. Sie dient dazu, die Leitsymptome zu erkennen und zu bewerten, Ausprägungen zu quantifizieren und pathologische Veränderungen im Einklang mit dem Leidensdruck des Patienten zu priorisieren.
Nach der freien Befragung des Patienten sind Leitsymptome abzufragen und in ihrem Verlauf einzustufen (Zeitdauer, Intensität, evtl. Progredienz) (Infobox 1).
-
Orbitale Leitsymptome
-
Exopthalmus/Enopthalmus
-
Ptosis/Lidretraktion
-
Augenbewegungsstörungen/Diplopie
-
periokuläre/okuläre Rötung und Schwellung
-
periokuläre Stauungszeichen/Chemosis
-
periorbitale/orbitale Schmerzen
-
Fremdkörpergefühl/Epiphora
-
retrobulbäres Druckgefühl
-
Bulbusbewegungsschmerz
-
Bulbusrepulsionsschmerz / erschwerte Repulsion
-
Augendruckanstieg
-
Aderhautfalten/Papillenödem
-
Gesichtsfeldausfall
-
Visusabfall
Danach ist es unverzichtbar, eine Anamnese zu allgemeinen Grunderkrankungen sowie eine Sozial- und Familienanamnese anzuschließen. Dabei ist der Fokus auf Autoimmunerkrankungen (z. B. Autoimmunthyreopathie), andere chronisch-entzündliche Erkrankungen (z. B. Granulomatose mit Polyangiitis), Malignome (z. B. Prostata-, Mamma-, Bronchialkarzinom, inkl. bestehender genetischer Prädispositionen) und die aktuelle Medikation (u. a. Antikoagulanzien) zu legen. Bekannte Vorerkrankungen der Orbita und der Nachbarstrukturen (Nase/Nasennebenhöhlen/Neurokranium) können einen Anhalt für Rezidiverkrankungen geben.
Da Erkrankungen der Orbita u. a. zu einem Verlust des Sehvermögens führen können und sich häufig primär ophthalmologisch manifestieren, ist die augenärztliche Anamnese von größter Bedeutung. Dabei muss darauf geachtet werden, ob beide Augen funktionell gleichwertig sind, ob eine Schwachsichtigkeit (Amblyopie) besteht, ob es Voroperationen im Bereich der Augen gab und wie sich die Sehschärfe und die Refraktion (Brechkraftverhältnisse) im Rahmen der möglichen Orbitaerkrankung entwickelt haben. Das Leitsymptom „Exophthalmus“ kann hier mit einem Pseudoexophthalmus (z. B. bei Oberlidretraktion) oder mit einem Enophthalmus der Gegenseite verwechselt werden.
3.2 Ophthalmologische Diagnostik [1]
-
Inspektion, Palpation, Bestimmung der Lidspaltenweite, Prüfung der Reponierbarkeit der Augäpfel, Testung der Hornhautsensibilität und des Augeninnendrucks
-
Bestimmung von Visus einschließlich Refraktion
-
Perimetrie: Gesichtsfelduntersuchung mit Bestimmung der Reizschwelle und Vermessung der Gesichtsfeldgrenzen und des blinden Flecks
-
Exophthalmometrie, Orthoptik
-
Spaltlampenuntersuchung, Ophthalmoskopie mit Beurteilung der Papille
Für detaillierte Ausführungen zur ophthalmologischen Anamnese und den Untersuchungtechniken möchten die Autoren auf einen aktuellen CME-Artikel zu diesem Thema verweisen [2].
3.3 Radiologische Diagnostik (MRT, CT, Sonografie)
Radiologische Verfahren ermöglichen
-
eine differenzialdiagnostische Einordnung
-
die Bestimmung der Ausdehnung
-
die Darstellung einer etwaigen Infiltration umgebender Strukturen
-
die OP-Planung (CT/MRT für Navigation)
-
die Bestrahlungsplanung
Die Sonografie kann erste Hinweise über die Art der Raumforderung geben, ist jedoch hinsichtlich der Tiefenausdehnung limitiert.
Die MRT ist der CT bei unklaren entzündlichen Läsionen und Tumoren überlegen. Es werden T1-, T2-gewichtete und T2-fettgesättigte Sequenzen (welche den Nervus opticus und die Augenmuskeln besonders gut zur Darstellung bringen) durchgeführt. Die zusätzliche Gabe von Kontrastmittel ist bei der Frage nach Tumoren und entzündlichen Prozessen sinnvoll.
Die CT erlaubt eine bessere Darstellung verkalkter und knöcherner Strukturen bei ausreichender Aussagekraft für Weichteilstrukturen und spielt daher bei der OP-Planung eine wichtige Rolle. Bei unklaren Raumforderungen und Entzündungen kann die Gabe von Kontrastmittel Zusatzinformationen hinsichtlich der Gewebeart und der Vaskularisation liefern. Ein Nachteil ist die Strahlenexposition, welche die Augenlinse schädigen kann und daher insbesondere bei jungen Patienten zu berücksichtigen ist.
Angiographie, Szintigraphie, PET und SPECT sind Spezialfällen vorbehalten. Für die Dokumentation und Planung von Rekonstruktionen sind auch die Fotografie, 3D-Fotografie und ggf. Oberflächenscans von Bedeutung [3].
3.4 Histopathologische Diagnostik
Kann mit den genannten Untersuchungstechniken keine suffiziente klinische Diagnose gestellt werden, ist die bioptische Sicherung von Gewebe mit histopathologischer Befundung von großer Bedeutung und insbesondere bei unklaren oder fortgeschrittenen Fällen, bei Rezidiven oder Therapieversagern sinnvoll. Hierbei sollten die Risiken durch iatrogene Verletzungen nicht höher sein als der erwartete Benefit. Für eine gezielte Planung der Probengewinnung sind eine adäquate klinische Untersuchung und Bildgebung mit anschließender interdisziplinärer Diskussion angeraten. Folgende Empfehlungen sollten hinsichtlich der Biopsieentnahme berücksichtigt werden:
-
möglichst keine Einnahme von systemischen Kortikosteroiden und anderen Immunsuppressiva in den Wochen vor der Biopsieentnahme
-
Entnahme inzisionaler oder exzisionaler Biopsien für Formalinfixierung und Untersuchungen an Frischmaterial
-
Entnahme ausreichend großer Biopsate; morphologische Untersuchungen an Feinnadelbiopsaten nicht immer möglich (nur bei Lymphomen oder Metastasen ausreichend)
-
Probenentnahme aus unterschiedlichen Arealen der Läsion
-
Vermeiden von Beschädigungen des Untersuchungsmaterials
-
Vergleich mit Vorbiopsien anderer Organe zur besseren Identifikation systemischer Erkrankungen
3.5 Labordiagnostik
Für eine Vielzahl seltener Erkrankungen der Orbita sind weitere Untersuchungen, insbesondere eine adäquate Labordiagnostik sinnvoll. Aufgrund des Umfangs der Arbeit möchten die Autoren diesbezüglich auf folgende Quelle verweisen: Laboratory Serologic Investigations; Fay, Aaron; Dolman, Peter J. Diseases and Disorders of the Orbit and Ocular Adnexa E-Book. Elsevier Health Sciences. S. 57-77 [4].
In den folgenden Kapiteln sollen nun die verschiedenen selten auftretenden strukturellen, entzündlichen, degenerativen und neoplastischen Veränderungen der Orbita systematisch dargestellt werden.
4. Strukturelle Läsionen der Orbita
Hierzu gehören angeborene und erworbene Veränderungen. Erstere beruhen auf Hamartomen und Choristomen, Teratomen und Gewebsektopien. Weiterhin zählt die große Gruppe der knöchernen Anomalien dazu. Bei den erworbenen Veränderungen stehen postinflammatorische und posttraumatische Zustände im Vordergrund.
4.1 Zysten und zystische Läsionen
Zystische Veränderungen können isoliert und multipel vorkommen. Zumeist wird eine mehr oder weniger kreisförmige Gestalt vorgefunden. Der Inhalt kann unterschiedlicher Konsistenz sein (serös, talgig, solide, gemischt etc.). In der Regel handelt sich um schmerzlose Raumforderungen, welche nur langsam an Größe zunehmen. Im Rahmen sekundär entzündlicher Reaktionen kann eine örtliche granulomatöse Reaktion des Gewebes stattfinden, welche zu starken reaktiven Veränderungen und im Bereich von Knochenstrukturen zu Osteolysen führen kann.
Allgemeine Einteilungsmöglichkeiten:
-
Zeitpunkt der Manifestation
-
angeboren
-
erworben
-
Anzahl
-
isoliert
-
multipel
-
-
Gewebetyp
-
epithelial
-
nicht epithelial
-
-
Lokalisation
-
subperiostal
-
extrakonal
-
konal
-
intrakonal
-
-
Ätiologie
-
vaskulär/hämatogen
-
neurogen
-
infektiös
-
metastatisch
-
4.1.1 Angeborene Zysten und zystische Läsionen epithelialen Ursprungs: Dermoide [5] [6] [7] [8]

-
Epidemiologie:
3–9 % der orbitalen Raumforderungen
-
Ätiologie:
kongenital als Choristom
-
Lokalisation:
-
meist extraorbital (präseptal) in temporal-superiorer Lokalisation, entlang der Sutura frontozygomatica (70–90 %) ([Abb. 1])
-
weniger häufig im Bereich des medialen Oberlids
-
selten als intraorbitale Dermoide vorliegend
-
Klinik:
-
gut abgrenzbare zystische Läsion mit langsamer Größenzunahme
-
Verdrängung angrenzender Strukturen
-
sonografisch gut abgrenzbare, echoarme, ovaläre bis runde Läsionen mit hyperreflektiven Binnenechos als Zeichen der epidermalen Differenzierung (Adnexstrukturen, Talg, Zelldetritus) oder sekundärer Kalzifikationen
-
fibröse Kapsel
-
Cave: rupturierte Dermoidzytsen induzieren eine granulomatöse Entzündung; akute und chronische Verläufe sind bekannt; auch ein transsuturales Wachstum ist möglich (Sanduhrkonfiguration)
-
Therapie:
-
komplette Exzision unter Erhalt der Zystenwand; insbesondere der proliferationsaktive Bereich (meist nahe des Knochens) ist zu entfernen
-
Marsupialisation
-
Cave: inkomplette Exzisionen können – wenn auch selten – zu Rezidiven führen. Noch seltener sind maligne Entartungen im Sinne von Plattenepithelkarzinomen.
-
Wichtige Differenzialdiagnosen:
-
temporal:
primäre und sekundäre Tumoren der Tränendrüse
-
medial:
-
Retentionszysten und Mukozelen (Anamnese bzgl. Knochenverletzungen)
-
Meningozelen; Enzephalozelen
-
Choristome der Nasenschleimhaut
-
generell:
jede solide, nicht infiltrative Raumforderung mit zystischer Gestalt
-
weitere zystische Veränderungen stellen konjunktivale Zysten, Epidermoide und Zysten, ausgehend von Gewebsektopien (z. B. ektopes Gewebe der Rathke-Tasche), Cholesterolgranulome und Cholesteatome dar.
Wird eine zystische Struktur innerhalb eines Gewebes vorgefunden (z. B. im äußeren Augenmuskel), ist von einem sekundären Geschehen (Infektion, Metastase) auszugehen.
4.1.2 Erworbene Zysten und zystische Läsionen
4.1.2.1 Mukozelen [9] [10] [11]

-
Epidemiologie:
-
3–4 % der orbitalen Raumforderungen
-
Altersgipfel 4. bis 6. Lebensdekade
-
Ätiologie:
-
fehlende Belüftung des betreffenden Sinus paranasalis
-
in 50 % gehen Traumata/Operationen voraus
-
gehäuftes Vorkommen bei Kindern mit zystischer Fibrose
-
Lokalisation:
-
medial bis medial-superior in der Orbita, entsprechend ausgehend vom Sinus ethmoidalis bzw. Sinus frontalis
-
selten ausgehend von der Keilbeinhöhle
-
subperiostal bis diffus intraorbital als Komplikation im Sinne einer Orbitalphlegmone
-
Klinik:
-
Exophthalmus/Bulbusverlagerung, Diplopie ([Abb. 2])
-
Schmerzen nur im Rahmen sekundär entzündlicher Veränderungen, ansonsten oft zunächst schmerzlos
-
verdrängender Charakter, wobei es sekundär zu Kompressionen des Nervus opticus, des Bulbus oculi oder der ableitenden Tränenwege kommen kann
-
Cave: Eine seltene Komplikation kann ein Enophthalmus sein. Im Rahmen eines „Silent-Sinus-Syndroms“ der Kieferhöhle ist eine Osteolyse des Orbitabodens möglich. Bei chronischen Verläufen kann eine Optikusatrophie resultieren.
-
Therapie:
-
Herstellung einer adäquaten Belüftung des betroffenen Sinus
-
ggf. Exzision und Obliteration
-
Komplikationsmanagement (Inzision, Drainage, intravenöse Antibiotikagabe)
Bei frontoethmoidaler Lokalisation ist an Meningozelen/Enzephalozelen zu denken. Ein nebenbefundlich bestehender Hypertelorismus und eine Größenzunahme während eines Valsalva-Manövers geben Hinweise darauf
CaveAuch bilaterale frontoethmoidale Mukozelen induzieren einen Hypertelorismus.
4.1.2.2 Dakryops [12] [13]

-
Epidemiologie:
-
0,5–2 % der orbitalen Raumforderungen
-
2. bis 5. Lebensdekade
-
Ätiologie:
-
Okklusion eines Drüsenausführungsganges (postentzündlich/posttraumatisch)
-
multifaktoriell
-
idiopathisch
-
Lokalisation:
-
meist Pars palpebralis der Tränendrüse (weniger häufig: Pars orbitalis, akzessorische Tränendrüsen, Zysten in ektopem Tränendrüsengewebe oder Zysten von Drüsengewebe der Karunkel) ([Abb. 3])
-
isoliert vorkommend, multiples Vorliegen und Bilateralität selten möglich
-
Klinik:
-
mehr oder weniger ausgeprägte Paragraphenform des Oberlides (temporal betonte Pseudoptosis)
-
Fremdkörpergefühl
-
schnelle Größenzunahme im Rahmen von emotionaler bzw. reflektorischer Tränensekretion oder infolge von Einblutungen (Farbveränderung) möglich
-
in Abhängigkeit von der Größe Auftreten von Astigmatismus und/oder Diplopie
-
Cave: selten sekundäre bakterielle Besiedlungen
-
Therapie:
-
komplette Exzision der Zyste(n)
-
Marsupialisation, Laser-Photokoagulation
-
Beobachten, Tränenersatzmittel, Meiden von kaltem Wind/Wasser
-
Wichtige Differenzialdiagnosen:
selten Dakryops als Folge eines Trachoms, eines Pemphigoids oder eines Tränendrüsensteins
4.1.2.3 Orbitale Implantationszysten [14] [15]
-
Epidemiologie:
heterogene Angaben in Abhängigkeit von der Ätiologie (am häufigsten nach Enukleation mit einem Auftreten in 2–7 % der Fälle)
-
Ätiologie:
posttraumatisch (Strabismuschirurgie, peri- und intraokuläre Chirurgie, penetrierendes Orbitatrauma, Enukleation, Orbitafraktur etc.) durch Aussaat von (konjunktivalem) Epithel
-
Klinik:
-
langsam größenprogredienter zystischer Tumor
-
Cave: Eine Ruptur führt zu granulomatösen Entzündungen.
-
Therapie:
komplette chirurgische Exzision
4.2 Neurogene Zysten
Die seltenen Anomalien beruhen auf angeborenen Störungen des Auges, des Sehnervs oder der Meningen.
4.2.1 Kongenitales Zystenauge [16] [17] [18]
-
Synonym:
Anophthalmus mit Orbitazyste
-
Epidemiologie:
sehr selten vorkommend
-
Ätiologie:
-
Folge einer fehlenden Invagination der primären Augenblase
-
multifaktoriell
-
Lokalisation:
-
zentral bis superior in der Orbita (Oberlid vorgewölbt)
-
Klinik:
-
meist isoliert, selten syndromal (Orbeli-Syndrom)
-
Zyste evtl. durch Neuroglia ausgekleidet
-
fehlender Bulbus oculi
-
kontralaterales Auftreten eines Mikrophthalmus möglich
-
zumeist mit systemischen Pathologien assoziiert:
-
Balkenagenesie, basale Enzephalozele, Heterotopie der grauen Substanz
-
Gesichtsspalten, Sattelnase, Choanalatresie, Keilbeinanomalien
-
Genitalfehlbildungen
-
Fingerfehlbildungen
-
Cave: Verbindung zum Subarachnoidalraum möglich
-
-
Therapie:
-
je nach Größe der Zysten Indikation zur chirurgischen Exzision
-
bei kleinen, größenkonstanten Zysten Beobachtung gerechtfertigt
-
bei zu großem chirurgischem Risiko evtl. Zystenpunktion erwägen
-
Wichtige Differenzialdiagnosen:
Teratome, die neben dem zystischen Anteil eine solide Komponente aufweisen und eine (bisweilen rasche) Größenprogredienz zeigen
4.2.2 Mikrophthalmus, Mikrophthalmus mit Zyste und Anophthalmus [16] [17] [18] [19] [20] [21] [22] [23] [24]
-
Epidemiologie:
-
Anophthalmus: 1–4 : 100.000
-
Mikrophthalmus: 2–20 : 100.000; 2–5% davon mit zusätzlichen Zysten
-
Mikrophthalmus mit Zyste in 2% der zystischen orbitalen Läsionen vorzufinden
-
Ätiologie:
-
Folge einer unvollständigen Einstülpung der primären Augenblase
-
multifaktoriell
-
SOX2-Gen auf Chromosom 3 bei Anophthalmus; weitere Gene (Auswahl): PAX6, OTX2, CHX10, FOXE3 und RAX
-
Mikrophthalmus/Anophthalmus isoliert und im Rahmen von syndromalen Fehlbildungen vorkommend (> 30 % syndromal; > 57 % multiple kongenitale Fehlbildungen)
-
prädisponierende Faktoren:
-
Schwangerschaft (Vitamin-A-Mangel, Strahlenexposition)
-
Infektionen (z. B. Röteln)
-
Diabetes mellitus
-
Drogenabusus
-
Medikamente
-
Konsanguinität
-
-
Lokalisation:
-
unilateral : bilateral = 2 : 1 bei Anophthalmus
-
meist unilateraler Mikrophthalmus
-
Mikrophthalmus mit Zyste inferior lokalisiert und das Unterlid vorwölbend (vgl. Abgrenzung zum Zystenauge)
-
Einteilung für Mikrophthalmus/Anophthalmus ([Tab. 1], Infobox 2):
|
Bezeichnung |
Merkmale |
|---|---|
|
Reiner Mikrophthalmus (syn. Nanophthalmus) |
▪ Augapfel relativ symmetrisch zu klein, wobei die Sagittalachse vermehrt betroffen ist |
|
▪ extreme Hyperopie (bis + 25 dpt) und flache Hornhaut (Radius 5,9 mm); selten: myoper Langbau |
|
|
▪ stark reduzierte Sehschärfe |
|
|
▪ Amblyopie, Nystagmus, konkomitante Esotropie, Ptosis, Enophthalmus, Mikrokornea |
|
|
▪ mentale Entwicklungsverzögerung |
|
|
▪ sekundäre Komplikationen: Winkelblock, Katarakt, Amotio chorioideae et retinae |
|
|
Mikrophthalmus mit Kolobom und ggf. Bulbuszyste |
▪ kombiniert mit primären (Mißbildungen) und sekundären (degenerativen) Veränderungen |
|
▪ ausgeprägter Mikrophthalmus mit starker Kolobomformation bis hin zum Anophthalmus |
|
|
▪ Zystenbildung im Bulbus oder in der Orbita |
|
|
▪ Katarakt |
|
|
▪ Pseudogliom durch Faltenbildung der Netzhaut und Mesodermproliferation (Bulbus septatus) |
|
|
▪ persistierendes hyaloidales Gefäßsystem und Tunica vasculosa lentis |
|
|
▪ schwerste Fehlbildungen des Nervus opticus (Hypo- bis Aplasie; Atrophie zu einem dünnen gliösen Strang) |
|
|
▪ weitere Veränderungen: Sklerokornea, große (nach hinten verlagerte) Linse, zum Teil Aphakie, Aniridie, Hypo- bis Aplasie des Ziliarkörpers |
|
|
▪ sekundäre Veränderungen: Pseudogliomatose, Orbitalzyste, Entzündungsfolgen |
|
|
Mikrophthalmus ohne nachgewiesenes Kolobom |
▪ mit sonstigen Veränderungen in Form und
Bau des Auges |
|
Mikrophthalmus mit systemischer Manifestation |
▪ Dysplasia
oculo-dento-digitalis |
|
▪ Dyskranio-pygo-phalangie
|
Weiteres Vorkommen von Mikrophthalmus: Dyscephalia oculo-madibulofacialis Ullrich-Fremery-Dohna, Typ Francois, Dyszephalie nach Hallermann-Streiff, Dysplasia oculo-vertebralis van der Hoeve-Syndrom, Potter-Syndrom, Fanconi-Syndrom
-
einseitig vs. beidseitig
-
ohne oder mit anderen okulären Pathologien
-
ohne oder mit systemischer Manifestation
-
nicht syndromal oder syndromal
-
Klinik:
-
schwerwiegende Fehlbildungen und funktionelle Blindheit des Partnerauges in 12,5 % bei unilateralem Mikrophtalmus bzw. in 34,0 % bei unilateralem Anophtalmus
-
zerebrale Pathologien – vor allem Fehlbildungen im Bereich des Corpus callosum – bei bilateralem Anophthalmus (in 70% der Fälle), bei unilateralem Anophthalmus (20 %) und bei unilateralem Mikrophthalmus (12,5 %)
-
Assoziation zu Spaltbildungen
-
gemeinsames Auftreten mit verschiedenen internistischen Erkrankungen möglich (kardiovaskulär, pulmonal, renal, gastrointestinal)
-
seltene Assoziationen:
-
Waardenburg-Syndrom (Innenohrschwerhörigkeit)
-
kongenitale ektodermale Dysplasie
-
Therapie:
-
je nach Größe der Zysten Indikation zur chirurgischen Exzision
-
bei kleinen, größenkonstanten Zysten Beobachtung gerechtfertigt
-
bei zu großem chirurgischem Risiko evtl. Zystenpunktion erwägen
-
Erweiterung der hypoplastischen Orbita mit Volumenexpandern
-
wichtige Differenzialdiagnosen:
Teratome, Kryptophthalmus (Infobox 3) und Phthisis bulbi als degenerative (postinflammatorische) Veränderung (Bulbus quadratus)
Alle Graduierungen vom sehfähigen, stark hyperopen Mikrophthalmus bis hin zum Anophthalmus sind beschrieben. Die Orbita mit den Adnexstrukturen (Lider und äußere Augenmuskeln) können nahezu regelrecht entwickelt vorliegen. Zum Teil existieren mikroskopisch kleine Bulbusrudimente, sodass die Grenze zum Anophthalmus extrem schwer zu ziehen ist. Manche Autoren sprechen erst nach histologischer Aufarbeitung des Orbitainhaltes ohne Nachweis okulären Gewebes von einem (echten, kompletten) Anophthalmus. Deshalb wurde der Begriff des „klinischen Anophthalmus“ oder „inkompletten Anophthalmus“ geprägt.
Anophthalmus
-
Fehlen sämtlicher okulärer Gewebe
-
primärer Anophthalmus – weder Auge noch Orbita entwickelt
-
sekundärer Anophthalmus – lebensunfähige Fehlbildung in Kombination mit Fehlbildungen des Großhirns
-
degenerativer Anophthalmus – Augenanlage vorhanden, aber degeneriert
-
-
häufig hypoplastische und damit in der Größe deutlich reduzierte Orbita, bei erheblicher Fähigkeit der Orbita zur Selbstdifferenzierung
-
Agenesie
-
kompletter Anophthalmus, auch sekundär möglich
-
Anwesenheit mesodermaler Gewebe
-
Anwesenheit ektodermaler Gewebe
-
Anwesenheit meso- und ektodermaler Gewebe
-
-
assoziierte Fehlbildungen der Sehbahn und Balkenagenesie möglich
-
Schädelfehlbildungen, Meningozelen, Dermoide der Orbita, Taubheit
-
Fehlen der Augenlider in Kombination mit Mikrophthalmus/Anophthalmus
-
Orbita hypoplastisch
-
Brauenbehaarung unvollständig bis komplett fehlend
-
meist bilateral
-
wenn unilateral, dann schwere Fehlbildungen der Orbita und des Auges auf der Gegenseite
-
Fehlen des Orbitadaches, Fehlbildung des Keilbeines, Ohrmuschelanomalien, Syndaktylie, Spaltbildungen des Gesichts, Kehlkopfatresien, Analatresie, Genitalfehlbildungen, Meningoenzephalozelen, Hydrozephalus, persistierender Ductus craniopharyngealis (orbitale Zystenbildung aus Resten der Rathke-Tasche), Hypophysenstörungen, Nebennierenaplasie
4.2.3 Orbitale Zephalozelen [16] [25] [26]
-
Epidemiologie:
-
Zephalozelen insgesamt: 0,8–4 : 10.000; in > 70 % der Fälle okzipitale Lokalisation
-
insgesamt < 1 % der orbitalen Raumforderungen bei Kindern
-
Ätiologie:
-
fehlerhafte Separationen des Neuroektoderms vom Ektoderm
-
persistierende Verbindungen zwischen dem Neurokranium und der Orbita
-
Einteilung:
-
anterior (Os frontale, Os ethmoidale, Os lacrimale, Maxilla)
-
posterior (Os sphenoidale)
-
Klinik:
-
Hypertelorismus, pulsierender Exophthalmus
-
schmerzloser (pulsierender) Tumor
-
Aplasie der Stirnhöhle
-
selten bilateral, dann mit Hypertelorismus vergesellschaftet
-
je nach Inhalt Unterscheidung zwischen Meningozelen (Hirnhäute) und Meningoenzephalozelen (Hirnhäute und Anteile des Gehirns)
-
weitere Schädelanomalien möglich (Keilbein), Hydrozephalus
-
Assoziation zur Neurofibromatose
-
Cave: sekundäre Ulzeration und Infektion der Zephalozele möglich
-
Therapie:
-
Exzision und Verschluss des ossären Defekts
-
Duraplastik
Prinzipiell liegen diese Läsionen bei Geburt vor, können sich aber mitunter erst im Laufe des Lebens manifestieren (v. a. wenn der Befund weit dorsal lokalisiert ist).
4.2.4 Andere Neurogene Zysten [27]
Primäre Arachnoidalzysten können in Kombination mit ipsilateralen okulären Kolobomen vorkommen. Als weitere Entität sind Nervenscheidenzysten mit anderen Anomalien des zentralen Nervensystems vergesellschaftet.
4.3 Angeborene Tumoren und Gewebsektopien
4.3.1 Dermolipom [28]

-
Epidemiologie:
etwa 2 % der orbitalen Tumoren im Kindesalter
-
Ätiologie:
-
Choristom
-
ektopes Ektoderm, welches als epidermales Gewebe der Lider im Bereich der Konjunktiva versprengt wurde und sich häufig nach intraorbital fortsetzt
-
Lokalisation:
-
temporaler Lidspaltenbereich
-
selten entlang der unteren und oberen Bindehautumschlagfalten
-
Klinik:
-
häufig asymptomatisch
-
Keratinisierung möglich
-
zum Teil Haarfollikel tragend
-
Fremdkörpergefühl
-
Therapie:
-
falls gewünscht (ästhetisch) bzw. bei Irritation der Augenoberfläche chirurgische Exzision unter Schonung von Bindehaut, Tränendrüse und äußeren Augenmuskeln (Musculus rectus lateralis)
-
oberflächliche Abtragung ohne Präparation in die Tiefe zur Orbita zumeist ausreichend
-
wichtige Differenzialdiagnosen:
-
Lymphom
-
orbitale Fetthernie ([Abb. 4])
4.3.2 Ektopes Tränendrüsengewebe und weitere Gewebsektopien [29] [30]
Akzessorisches Tränendrüsengewebe kommt regelmäßig im Bereich der Bindehautumschlagfalten vor (Wolfring- und Krause-Drüsen). In den posterioren Bereichen der Orbita ist dieses Gewebe jedoch selten zu finden. Hier kann es jedoch eine chronisch vernarbende Entzündung induzieren, was die chirurgische Sanierung erfordert.
Darüber hinaus wurden bereits heterotopes Hirngewebe und Knorpelstrukturen in der Orbita beschrieben.
4.3.3 Orbitales Teratom [31] [32] [33]
-
Epidemiologie:
-
6,6 % der pädiatrischen Tumoren, meist jedoch extraorbital gelegen
-
orbital sehr selten
-
Ätiologie:
Neoplasie aus zwei oder allen drei Keimblättern (pluri- bis totipotente embryonale Stammzellen)
-
Klinik:
-
größenprogredienter, unilateraler, zumeist zystischer Tumor
-
Proptosis
-
Vergrößerung der knöchernen Orbita um den Faktor 2 bis 3 möglich
-
stark heterogenes Gewebe enthalten:
-
komplettes Teratom als orbitaler Fetus-in-fetus (Orbitopagus parasiticus)
-
inkompletter zweiter Fetus mit Anteilen einer Wirbelsäule
-
Teratom mit Nachweis aller drei Keimblätter
-
Dermoidtumor mit Nachweis von zwei Keimblättern
-
-
Therapie:
chirurgische Exzision
-
wichtige Differenzialdiagnosen:
-
Dermoide, Rhabdomyosarkome oder vaskuläre Anomalien
-
Cave:
-
malignes Teratom der Orbita (in 2 % der Fälle)
-
intrakranielle Ausdehnung
4.4 Knöcherne Anomalien und mesodermale Defekte
4.4.1 Akzessorische Knochen und Suturen, ossäre Varianten, aberrierende Foramina [34] [35] [36]
-
Epidemiologie:
-
manche Formen häufig (Duplikatur der Sutura frontozygomatica in 7 von 400 Schädeln)
-
erhebliche regionale Unterschiede
-
in ca. 1–2 % der Augenhöhlen nachweisbar
-
Ätiologie:
-
Störung der Ossifikation
-
Unterteilung in Normvarianten und kongenitale Deformitäten
-
Orbita bzgl. der Entwicklung weitgehend selbstdeterminiert (unabhängig vom Auge)
-
Leitungsbahnen werden erst sekundär vom Knochen umschlossen; Nerven- oder Gefäßduplikaturen gehen somit der ossären Variante voraus
-
Klinik:
-
zum Teil klinisch vollkommen inapparent
-
Assoziation mit Dysostosen des Schädels
-
Exophthalmus
-
Gesichtsasymmetrie
-
Hyper- oder Hypotelorismus
4.4.1.1 Beispiele für knöcherne Varianten [34]
-
akzessorische Suturen und Knochenfragmente (z. B. Duplikatur der Sutura frontozygomatica, fragmentiertes Os zygomaticum mit einem Ossiculum infraorbitale marginale)
-
fehlende Beteiligung der Maxilla an der Bildung der inferioren Orbitakante (Häufigkeit: 1 : 2.250)
-
Fehlbildungen des Os ethmoidale, bei denen das Os frontale die mediale Orbitawand bildet
-
häufig Dehiszenzen der Lamina papyracea
-
inkomplettes Vorliegen bis vollständiges Fehlen des Os lacrimale; Variationen des Hamulus lacrimalis und damit Formvariationen des Saccus lacrimalis und des Ductus nasolacrimalis
-
akzessorische Knöchelchen im Bereich des Processus frontalis maxillae
-
Hypoplasie der Maxilla
-
Duplikatur der Crista lacrimalis anterior
4.4.1.2 Beispiele für Varianten der Foramina [34]
-
supra- und infraorbitale Foramina in Form von Kanälen/Inzisuren/Sulci, Mehrfachanlagen
-
Duplikatur des Canalis opticus / Fehlen des Foramen opticum bei Anophthalmus
-
bis zu vierfache Anlage des Foramen zygomaticofaciale
-
bis zu fünffache Anlage des Foramen infraorbitale (Doppelanlagen in > 10 %)
-
Spina trochlearis (einfach, zweifach oder als Ring)
-
ossäre Teilung der Fissura orbitalis inferior durch akzessorische Knochenspange
4.4.1.3 Beispiele für Wanddefekte [34]
-
unilaterale Nichtausbildung des Os frontale und der Maxilla (Frontallappen nur durch Meningen und Haut bedeckt; klinisch auch ohne Vorliegen einer Zephalozele bei Bulbustiefstand und Divergenz)
-
Cave: Assoziation zu Zyklopie und Arhinenzephalie, selten Koinzidenz mit Phakomatosen, z. B. bei Neurofibromatose (Fehlbildungen im Bereich des Os sphenoidale)
4.4.1.4 Beispiele für Wanddehiszenzen [34]
-
infantiles und bisweilen seniles Auftreten physiologisch
-
vor allem im Bereich von Maxilla und Os ethmoidale
-
seltener im Bereich von Os lacrimale, Os palatinum oder Os sphenoidale
-
Cave:
-
Variation des Canalis opticus mit Fehlen von Anteilen der knöchernen Umschließung des Nervus opticus und daraus resultierender direkter Kommunikation zwischen Durascheide und Keilbeinschleimhaut
-
Assoziation orbitaler Varizen mit knöchernen Dehiszenzen bzw. Fehlinterpretation von Phleboliten als solche möglich
4.4.2 Cribra orbitalia [34] [35]
Ausdünnung des Orbitadaches mit freiliegender Diploë und darin befindlichem Venenplexus als Folge einer Knochenentwicklungsstörung (DD: Vitamin-D-Mangel, Anämie)
4.4.3 Familiäre Hypoplasie des Orbitarandes (Urrets-Zavalia-Syndrom) [34]
-
Epidemiologie:
extrem selten (< 1 : 1.000.000; bisher in zwei Familien beschrieben)
-
Ätiologie:
-
genetisch bedingte Agenesie von Orbitakante und Teilen der okulären Adnexstrukturen
-
dominanter Erbgang (hohe Penetration, konstante Expression)
-
Gewebe des paraxialen und viszeralen Mesoderms betreffend
-
Klinik:
-
Fehlen des knöchernen Orbitarahmens
-
Hypoplasie der Lidhaut und der Tarsus palpebrales
-
variable Defekte mit Störungen der ableitenden Tränenwege, Lidkolobom, Strabismus verticalis
Oftmals haben akzessorische Knochen und Suturen bzw. aberrierende Foramina keinerlei Krankheitswert, zum Teil müssen sie aber im Rahmen von Sinus-, Orbita- und Tränenwegschirurgie berücksichtigt werden.
4.4.4 Gesichtsspalten, faziale Dystrophien, kraniofaziale Dysostosen und Schädelfehlbildungen [34] [37] [38]
Komplexe Gesichtsdeformitäten, die auf einer Störung der Embryogenese im Bereich des 1. (und 2.) Kiemenbogens beruhen.
4.4.4.1 Seltene Gesichtsspalten (u. a. Tessier-Spalten) [37] [38] [39]
-
Epidemiologie:
1–5 : 100.000
-
Ätiologie:
-
fehlende Fusion der embryonalen Gewebe des 1. Kiemenbogenpaares (Assoziation zu Mittelohrfehlbildungen von Hammer und Amboss)
-
frühes Entwicklungsstadium (5. bis 8. Embryonalwoche)
-
in späteren Stadien durch Nabelschnurtrauma (atypische Spalte; „Amniotic-band-syndrome“)
-
typische Form:
-
oro-okuläre bzw. oro-orbitale Gesichtsspalte
-
transversale und schräge Gesichtsspalten
-
Klinik:
-
zahlreiche Ausprägungen
-
zum Teil mediale Lidkolobome
-
-
atypische Form:
-
oro-temporale Gesichtsspalte
-
Klinik:
-
temporale Lidkolobome
-
Assoziation zu Dermoiden, Ohranhängseln, anderen Gesichtsdeformitäten (Lippen-Kiefer-Gaumenspalte)
-
Anophthalmus, Mikrophthalmus, Uvealkolobom
-

-
15 verschiedene Arten (0 bis 14), Verlauf vom Oberkiefer über die Orbita bis zum Os frontale möglich
-
Gruppen:
-
Mittellinienspalten
-
Paramedianspalten
-
Orbitalspalten
-
Seitenspalten ([Abb. 5])
-
Therapie:
-
plastische Rekonstruktion durch MKG-Chirurgie zum Verschluss des Weichteil- und Knochendefektes; kieferorthopädische Behandlung
-
phoniatrische/pädaudiologische Anbindung
-
ggf. okuloplastische Intervention zur Lidstellungskorrektur oder zum Kolobomverschluss
4.4.4.2 Faziale Dystrophien, kraniofaziale Mikrosomie [37] [41]
-
Epidemiologie:
1 : 3.000 bis 1 : 5.000
-
Ätiologie:
Störungen der Embryogenese (den 1. Kiemenbogen betreffend)
-
Klinik:
-
Kieferknochen:
-
asymmetrische Mittelgesichtshypoplasie
-
Ankylose
-
Mikrognathie
-
-
Auge:
-
Dermoide (epibulbär)
-
Höher-/Tieferstand
-
Anophthalmus/Mikrophthalmus
-
Kolobome
-
-
Mundregion:
-
Makrostomie (Spalten)
-
Gesichtsspalten
-
-
Skelettsystem:
Wirbelsäulenveränderungen
-
Hirnnerven:
-
Fazialisparese
-
Schallempfindungsstörung
-
Gaumenhebungsstörung
-
Augenmuskelparese
-
-
Ohr:
-
Ohranhängsel
-
Mikrotie/Anotie
-
Gehörgangsatresie
-
Schallleitungsstörung
-
-
Cave: seltene Assoziationen mit Fallot-Tetralogie, Ventrikelseptumdefekt, Transposition der großen Gefäße, Aortenbogenanomalien, Nierenfehlbildungen (Fehlen einer Niere, Doppelanlage des Ureters, Nierenektopien), Hydronephrose, Hydroureter, Anomalien der Extremitäten (Elle, Speiche), Mikrozephalie, Enzephalozele, Hydrozephalie, Balkenhypoplasie, Arnold-Chiari-Malformation, Holoenzephalie
-
als Syndrom vorliegend: VA(C)TER(L) – vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal atresia, renal anomalies and limb anomalies; CHARGE – coloboma, heart, atresia choanae, retardation of growth and development and genitourinary and ear anomalies
-
Formen ([Tab. 2]):
|
Bezeichnung |
Beschreiber |
Lateralität |
Merkmale |
|---|---|---|---|
|
Otozephalie |
St. Hilaire |
bilateral |
|
|
▪ Aplasie bzw. schwere Hypoplasie der Mandibula |
|||
|
▪ Deformität von Zunge, Nase und Ohren |
|||
|
▪ transversale Gesichtsspalte |
|||
|
▪ Augenfehlbildung (Anophthalmus) |
|||
|
Mandibulo-faziale Dysostose |
Franceschetti, Zwahlen, Klein, Treacher-Collins |
bilateral |
▪ Lidkolobom (nach temporal abfallende Lidachse) |
|
▪ hypoplastische Gesichtsknochen (Maxilla/Mandibula) |
|||
|
▪ Fehlbildungen des äußeren Ohres und z. T. des Mittelohres |
|||
|
▪ Makrostomie, hoher Gaumen |
|||
|
▪ Fistel zwischen Ohr und Mund |
|||
|
▪ zur Wange verlagerter Haaransatz |
|||
|
▪ Gesichtsspalten, Skelettdeformitäten |
|||
|
▪ selten: inkomplette und unilaterale Fälle |
|||
|
Oto-mandibuläre Dysostose |
Francois und Haustrate |
unilateral |
▪ Agenesie/Aplasie der Mandibula |
|
▪ Malformationen des Kiefergelenks |
|||
|
▪ Fehlbildungen des äußeren Ohres |
|||
|
▪ Gehörgangsatresie |
|||
|
▪ Ohranhängsel |
|||
|
▪ Fisteln zwischen Mund und Tragus |
|||
|
▪ transversale Gesichtsspalte |
|||
|
▪ Lippen-Kiefer-Gaumenspalte |
|||
|
▪ Zahnfehlstellungen |
|||
|
Okulo-aurikuläre Dysplasie |
Goldenhar |
unilateral |
▪ epibulbäre Dermoide |
|
▪ Ohranhängsel |
|||
|
▪ Ohrfisteln |
|||
|
▪ Kieferhypoplasie (vor allem der Mandibula) |
|||
|
▪ Kolobom des Oberlides |
|||
|
▪ Makrostomie |
|||
|
▪ Mikrophthalmus |
|||
|
▪ nervale, muskuläre und skelettale Anomalien |
|||
|
Mandibulo-okulo-faziale Dyszephalie |
Hallermann und Streiff; Ullrich und Fremerey-Dohna |
unilateral |
▪ Kieferhypoplasie (v. a. der Mandibula) |
|
▪ Nasenanomalie |
|||
|
▪ kongenitaler Katarakt, Mikrophthalmus, Mikrokornea |
|||
|
▪ Kleinwuchs |
|||
|
▪ Zahnanomalien |
|||
|
▪ Hpotrichose (Brauenbehaarung) |
|||
|
▪ Hautatrophie |
|||
|
Okulo-vertebrale Dysplasie |
Weyers und Thier |
unilateral |
▪ Dysplasie der Maxilla |
|
▪ Gesichtsasymmetrie |
|||
|
▪ Deformation des maxillären Alveolarfortsatzes |
|||
|
▪ Okklusionsstörungen |
|||
|
▪ Anophthalmus, Kryptophthalmus |
|||
|
Mikrogenie und Glossoptosis |
Pierre Robin |
▪ Hypoplasie der Mandibula |
|
|
▪ Glossoptose |
|||
|
▪ Gaumenspalte |
|||
|
▪ Netzhautablösung |
|||
|
▪ Katarakt |
|||
|
▪ Mikrophthalmus |
|||
|
▪ Glaukom |
|||
|
▪ hohe Myopie |
-
Otozephalie
-
mandibulo-faziale Dysostose
-
oto-mandibuläre Dysostose
-
okulo-aurikuläre Dysplasie
-
mandibulo-okulo-faziale Dyszephalie
-
okulo-vertebrale Dysplasie
-
Mikrogenie und Glossoptosis
4.4.4.3 Faziale (Hemi-)Atrophie (Parry-Romberg) [42] [43]
-
Synonym:
progressive faziale Trophoneurose
-
Epidemiologie:
-
1 : 700.000
-
Frauen häufiger als Männer betroffen
-
Ätiologie:
-
uni- oder selten bilaterale Störung des Weichgewebes und Knochens durch ausbleibende/insuffiziente Innervation (Hirnnerven V und VII)
-
Klinik:
-
langsam fortschreitende, in der Regel einseitige Atrophie der Gesichtsweichteile, einschließlich der Muskulatur, sowie teilweise der knöchern-knorpeligen Strukturen
-
Gesichtsasymmetrie
-
sehr variable Krankheitsaktivität über 2 bis 20 Jahre bis zum selbstlimitierenden Stillstand
-
Fehlbildung des äußeren Ohres (Tieferstand, Gehörgangsatresie, Anhängsel, verkleinerte Gehörknöchel, kleine Paukenhöhle)
-
verkleinerter Ober- und Unterkiefer (unilaterale Mikrognathie)
-
hypoplastisches Mastoid
-
Atrophe Gesichtsmuskulatur, gelbliche Verfärbung der Haut (Hypo- und Hyperpigmentationen möglich), Lanugobehaarung bei sonstiger Alopezie
-
temporal abfallende Lidachse, Enophthalmus, Lagophthalmus, Pseudoptosis (DD: assoziiertes Horner-Syndrom)
-
freiliegender Nervus facialis im Mittelohr
-
Atrophie von weichem Gaumen und Zunge (Sprech- und Schluckstörungen)
-
säbelförmige Sklerodermie entlang der Haut der medialen Orbita
-
Heterochromie, Blepharophimose, Kolobom, Okulomotoriusparese, zum Teil Nystagmus
-
Cave:
-
selten Hemiatrophie des gesamten Körpers
-
häufigeres Auftreten orbitaler Tumoren (u. a. Neurinome)
-
Therapie:
-
systemisch medikamentös (u. a. Glukokortikoide, Cyclophosphamide, MTX) und lokaltherapeutisch (u. a. PUVA, Botulinumtoxin, Phototherapie)
-
operative Therapieoptionen zur Stabilisierung oder Wiederherstellung bei periokulären Veränderungen
-
wichtige Differenzialdiagnosen:
-
faziale Hemihypertrophie:
-
-
unilaterale Hypertrophie einer Körperhälfte
-
Exophthalmus bei Megalophthalmus
-
kongenitale Fazialisparese (Möbius-Syndrom)
-
4.4.4.4 Kraniofaziale Dysostosen [44] [45] [46] [47] [48]
Bei den kraniofazialen Dysostosen handelt es sich um Erkrankungen mit heterogener Befundausprägung.
-
Epidemiologie:
-
syndromal und nicht syndromal
-
3–10 : 10.000
-
syndromale Formen sind um den Faktor 10 bis 50 seltener; bestimmte Formen extrem selten mit < 1 : 1.000.000 (Boston-Syndrom)
-
häufigste Syndrome:
-
Crouzon-Syndrom
-
Pfeiffer-Syndrom
-
Saethre-Chotzen-Syndrom
-
seltene Syndrome:
-
Boston-Syndrom
-
Cole-Carpenter-Syndrom
-
Herrmann-Opitz-Kraniosynostose
-
Kraniosynostose Typ Philadelphia
-
Kardio-kraniales Syndrom Typ Pfeiffer
-
Jackson-Weiss-Syndrom
-
Hunter-McAlpine-Kraniosynostose
-
Lopez-Hernandez-Syndrom
-
Baller-Gerold-Syndrom
-
Ätiologie:
-
Anlagestörung der Knochen, vorzeitiger Verschluss betreffender Suturen
-
durch expansives Hirnwachstum und Blockade in einer Richtung resultierende vermehrte Volumenexpansion in einer anderen Richtung (Virchow-Regel: Blockade senkrecht zur Synostose und kompensatorisches Wachstum entlang der Synostose)
-
Sutura metopica (physiologisch bis zum 8. Lebensmonat offen), Sutura sagittalis, Sutura lambdoidea, Sutura coronalis nicht bis zum Erwachsenenalter offen
-
Manifestation der Erkrankungen typischerweise im 1. bis 4. Lebensjahr
-
Klinik:
-
Schädeldeformität mit oder ohne Orbitabeteiligung
-
Exophthalmus, Hyper- und Hypotelorismus, Strabismus
-
erhöhter Hirndruck (Kopfschmerzen, Papillenödem, Optikusatrophie)
-
Enzephalozelen
-
Syndaktylien
-
Ohrfehlbildungen (Ohrmuschel, äußerer Gehörgang, Mittelohr)
-
Assoziationen zu Stoffwechselstörungen (Hyperthyreose, Vitamin-D-resistente Hypophosphatämie, Mukopolysaccharidose [Def.: früher als Gargoylismus bezeichnete Dysmorphie des Kopfes] und Mukolipidose), Chondro- und Osteodystrophien (z. B. Achondroplasie)
4.4.4.4.1 Dysostosis cleidocranialis [44] [49]
-
hypoplastische Dysostose des Schädels mit Brachy- oder Platyzephalie ([Abb. 6])
-
Zahnanomalien
-
Pseudoarthrosen, Hypoplasien oder Aplasien einer oder beider Schlüsselbeine
-
Hypoplasie des Os nasale und der Maxilla
-
hoher Gaumen
-
Deformitäten von Thorax, Becken und Extremitäten möglich
-
Spina bifida

4.4.4.4.2 Oxyzephalie (Akrozephalie, Turmschädel) [44] [45] [46] [47] [48]
-
Geschlechterverteilung: 4 : 1 (Männer : Frauen)
-
vertikale Elongation des Schädels
-
transversale und sagittale Verkürzung
-
koronare und faziale Synostosen
-
Verkleinerung der Schädelbasis
-
prominente Nase, hypoplastischer Oberkiefer, enger Gaumen
-
flache Orbitae mit geringen Volumina
-
supraorbital betonter Exophthalmus, Strabismus, Motilitätsstörungen, Nystagmus, Expositionskeratopathie, milder Hypertelorismus
-
Papillenödem, Optikusatrophie
-
retinale vaskuläre Stase, Tortuositas vasorum
-
Lidödeme
-
seltene Assoziationen: Katarakt, Hornhautdystrophien, Lidkolobom, orbitale Enzephalozele
-
oftmals leichte Intelligenzminderung (Cave: die Intelligenzminderung kann häufig Folge der psychosozialen Stigmatisierung sein und ist nicht organisch bedingt!)
-
klassisches Syndrom: Apert-Syndrom (Akrozephalo-Syndaktylie)
-
Oxyzephalie mit Exophthalmus, Expositionskeratopathie, Ophthalmoplegie, Papillenödem, Optikusatrophie
-
Syndaktylie (2. bis 4. Finger/Zehe); Synarthrosen (Schulter, Ellenbogen)
4.4.4.4.3 Skaphozephalie (Sphenozephalie) [44] [45] [46] [47] [48]
-
Synostose der Sutura sagittalis
-
lange, schmale Schädelform
-
geringe Breite des Schädels mit Hypotelorismus
-
sagittale Synostose
4.4.4.4.4 Brachyzephalie [44] [45] [46] [47] [48]
-
Synostose der Suturae coronales oder lambdoideae
-
kurzer, breiter Schädel
-
Zwischenform aus Skaphozephalie und Oxyzephalie
4.4.4.4.5 Dolichozephalie [44] [45] [46] [47] [48]
-
prominenter frontaler Schädel
-
coronale, sagittale und/oder lambdoide Synostosen
-
koronare Vertreter: Crouzon-Syndrom
-
prominente Stirn, typische Nasenform (Papageienschnabel)
-
hypoplastische Maxilla, Prognathie, Zahnfehlstellungen, hoher Gaumen, zum Teil mit Spaltbildung
-
divergenter Strabismus, Tränenwegsstenose, Papillenödem, Optikusatrophie, Expositionskeratopathie, okuläre Kolobome, Glaukom, Linsenektopie
-
Ohranomalien (Gehörgangsatresie, Ohrmuschelanomalie)
4.4.4.4.6 Plagiozephalie [44] [45] [46] [47] [48]
-
sagittale Asymmetrie
-
unilaterale Synostose verschiedener Schädelnähte
-
unterteilt in:
-
anteriore Form:
-
einseitige Synostose der Sutura coronalis
-
ipsilaterale Orbita hypoplastisch
-
Orbitadach mit nach temporal abfallender Achse
-
posteriore Form:
-
Verschluss der Sutura lambdoidea
-
differenzialdiagnostisch gegenüber einem erworbenen, lagerungsbedingten Plagiocephalus abzugrenzen
4.4.4.4.7 Hemikraniose [44] [45] [46] [47] [48]
-
prominentes Os frontale, Os parietale und Os zygomaticum
-
erhebliche Asymmetrie
-
Exophthalmus und Optikusatrophie
4.4.4.4.8 Trigonozephalie [44] [45] [46] [47] [48]
-
frontale Schädelunterentwicklung (Synostose der Sutura metopica) mit okzipitaler Kompensation
-
auch bei koronarer Synostose
-
Hypotelorismus
-
Optikusatrophie
-
schmale vordere Schädelgrube
-
hypoplastischer Sinus ethmoidalis
4.4.4.4.9 Platyzephalie (Klinozephalie) [44] [45] [46] [47] [48]
-
vergrößerte hintere Schädelgrube mit Überreiten der Halswirbelsäule
-
Synostose der superioren Nähte
-
Therapie:
-
konservativ über Änderung der Schädellagerung, mittels Physiotherapie bis hin zur Helmtherapie
-
Korrektur oder Prävention eines erhöhten Hirndrucks
-
Behandlung der Kopfdeformität (funktionelle/psychosoziale Indikation)
-
Operationstechniken:
-
offene oder minimal-invasive Suturektomie
-
Suturektomie und Entfernung bzw. Modellierung einzelner Knochenfragmente, ggf. als Frontoorbitales Advancement
-
kraniale Distraktion
4.4.4.5 Hypertelorismus [37] [44] [45]
-
Interorbitalabstand > 30 mm
-
Assoziation mit Kraniosynostose oder Spaltbildungen im Kiefer- und Gesichtsbereich, aber auch isoliert auftretend
-
selten Vorkommen von frontalen bzw. frontobasalen Enzephalozelen
-
Einteilung in:
-
primär:
-
morphogenetisch (habituell) durch dysproportionales Wachstum
-
embryonal durch Entwicklungsfehler (z. B. Apert- und Crouzon-Syndrom) mit Spaltbildung der Nase (selten doppelt angelegtes Septum nasi, Dermoid)
-
sekundär:
Störung der Entwicklung durch Umgebungsfaktoren (Trauma, Enzephalozelen, Erkrankungen des Knorpels oder der Knochen)
4.4.4.6 Mittellinienverschlussstörungen [50]
-
Synonyme/Ausprägungen:
Zyklopie, Holoprosenzephalie, Arhinzephalie, otozephale Anomalien
-
Epidemiologie:
1 : 16.000 Lebendgeborenen
-
Ätiologie:
-
komplexe zerebrale Fehlbildung mit ausbleibender Hemisphärentrennung
-
Zyklopie ist Folge eines gestörten Determinationsprozesses, der aus fördernden und hemmenden Abläufen besteht und die gesamte Kopfanlage betrifft
-
Klinik:
-
teilweise Vorliegen eines rüsselartigen Anhangs (Proboscis) als Rudiment des Nasenapparates
-
Fehlbildungen wie komplettes Fehlen der Augen (Anops) oder Fehlen des Gesichtes (Aprosposus – Augen-, Geruchs- und Geschmacksorgan fehlen) sowie Vorliegen von einer (Zyklopie bzw. Synophthalmie) über drei (Diprosopus triophthalmus, Zephalothoracopagus) bis zu vier getrennten Augenanlagen (Tetrophthalmus) möglich
-
beginnend vom Sehnerv getrennte Darstellung beider Augen und Orbitae von posterior nach anterior in zunehmendem Maße möglich (zum Beispiel mit einfacher Kornea bei ansonsten doppelt angelegten übrigen Geweben)
-
viele klinische Variationen mit fließenden Übergängen, z. B. mediane Gesichtsspalten, Mikrophthalmus, Anophthalmus, Zyklopie, Hypotelorismus (faziale Dysmorphie korreliert mit zerebraler Anomalie)
-
isoliert oder im Rahmen von Syndromen vorkommend (u. a. CHARGE, Smith-Lemli-Opitz-Syndrom, Rubinstein-Taybi-Syndrom, Meckel-Syndrom, Lambotte-Syndrom, Steinfeld-Syndrom)
-
Komplikationen:
-
neurologisch: Epilepsie, Hydrozephalus, mentale Entwicklungsverzögerungen, Hypotonie, Spastiken
-
kraniofazial: Mikrozephalie, Hpo- und Hypertelorismus, mediane und laterale Spalten (Lippen, Kiefer und Gaumen betreffend), Arhinie, Hypoplasie der Apertura piriformis
-
endokrinologisch: Diabetes insipidus, Wachstumshormonmangel, Nebennierenrindenhypoplasie, Hypogonadismus
-
oromotorische Dysfunktion: Sprech-, Schluckstörungen, gastroösophagealer Reflux, Notwendigkeit einer Gastrostomie
-
vegetative Dysfunktionen: Temperaturregulation, Herz- und Atemfrequenzsteuerung
-
-
Lebenserwartung:
Prognose ungünstig, in früher Kindheit bereits letal (mittleres Sterbealter bei 4 Jahren; 15 % zwischen 10 und 19 Jahren)
-
Therapie:
-
symptomorientiertes interdisziplinäres Komplikationsmanagement
-
4.4.5 Entwicklungsstörungen der Knochen
4.4.5.1 Fibröse Dysplasie [51]
-
Ätiologie:
mesenchymaler Defekt der Knochenspongiosa und des Knochenmarks und Ersatz durch fibröses Gewebe
-
Klinik:
-
Unterformen:
-
Befall nur eines Knochens (monoostotisch)
-
Befall mehrerer Knochen (polyostotisch)
-
McCune-Albright-Syndrom (schwerste Form mit Kleinwuchs, endokrinen Störungen und Pigmentstörungen der Haut)
-
Affektion aller Schädelknochen möglich
-
bei kranialer Lokalisation orbitale Beteiligung in > 40 % der Fälle
-
atypischer Gesichts- und Kopfschmerz
-
Sinusitis
-
Hörstörungen
-
Exophthalmus
-
Gesichtsasymmetrie
-
Therapie:
-
chirurgische Dekompression (Optikuskanal, Orbita) bei klinischen Beschwerden wie Optikusneuropathie
-
chirurgische Exzision betroffener Bereiche unter Berücksichtigung funktioneller Gesichtspunkte (kurative Chirurgie nicht möglich, prinzipiell gutartige Erkrankung)
-
Cave: Radiatio aufgrund des Risikos einer malignen Entartung kontraindiziert
4.4.5.2 Osteopetrose [52]
-
Epidemiologie:
1 : 250.000 bis 5 : 100.000 (bei dominantem Erbgang)
-
Ätiologie:
-
gestörte Differenzierung der Osteoklasten
-
Vermehrung der Knochensubstanz bei geminderter Stabilität
-
Klinik:
-
Makrozephalie, kraniofaziale Dysmorphie
-
Frakturen, Kleinwuchs
-
Nervenkompressionen (Blindheit, Taubheit, Fazialisparese)
-
Osteomyelitiden
-
Hydrozephalus, Hypophysenhypoplasie, zerebrale Demyelinisierung
-
Choanalatresie
-
Zahnanomalien, doppelte Zahnreihen, Gaumenspalte, Karies
-
Störung des Kalziumhaushalts, sekundärer Hyperparathyreoidismus
-
Retinaatrophie, Katarakt
-
Störung der Hämatopoese mit Gefahr der Panzytopenie, Hepatosplenomegalie
-
Ausprägungen, Formen:
-
autosomal-rezessive Osteopetrose (ARO) (maligne Form)
-
neuropathische Form der ARO
-
Assoziation mit renal-tubulärer Azidose (RTA)
-
„X-linked Osteopetrosis“ mit Lymphödem, anhidrotisch-ektodermaler Dystrophie und Immundefizienz (OLEDAID)
-
„Common variable immune deficiency“ (CVID)
-
„Leukocyte adhesion deficiency syndrome“ (LAD-III)
-
autosomal-dominante Form (Albers-Schönberg-Krankheit)
-
Pyknodysostose
-
Dysosteosklerose
-
Osteopoikilose (bei Vorkommen von Bindegewebsnävi als Buschke-Ollendorf-Syndrom)
-
Osteopathia striata (mit oder ohne kraniale Sklerose)
-
-
Therapie:
-
symptomatisch je nach Ausprägung der Funktionsstörung bzw. Komplikation (z. B. chirurgische Dekompression des Nervus opticus)
-
multi- und interdisziplinär
-
Vitamin-D-Substitution
-
ggf. Transfusion
4.4.5.3 Cherubismus [53]
-
Ätiologie:
-
unterschiedliche Erbmodi, wahrscheinlich vor allem dominant mit hoher Penetration
-
zystische Proliferation der Mandibula und Maxilla
-
Klinik:
-
Krankheitsbeginn im Kindesalter (2. bis 5. Lebensjahr)
-
unterschiedliche Verläufe, zum Teil klinisch nahezu inapparent
-
hervorstehendes Kinn und prominente Wangen, Störungen der Zahnstellung
-
Augen nach oben verdrängt, Proptosis, Diplopie
-
derbe Knochenschwellungen
-
bisweilen Assoziation mit regionaler Lymphadenopathie
-
spontane Involution nach Abschluss des Knochenwachstums (etwa um das 30. Lebensjahr)
-
Cave: Dysarthrie, Dysphagie und Dyspnoe möglich
-
Assoziation mit Ramon-Syndrom, Neurofibromatose 1 und Fragilem-X-Syndrom
-
Therapie:
-
chirurgische Resektion der befallenen Regionen unter funktionellen Gesichtspunkten
-
Cave:
-
Krankheitsaktivierung durch Chirurgie möglich
-
Radiotherapie kontraindiziert (Osteoradionekrose, Malignominduktion)
Spaltbildungen und andere kraniofaziale Fehlbildungen zeigen eine sehr große Variationsbreite. Sie können isoliert und syndromal in Erscheinung treten. Klinisch kann es dabei zum Teil komplizierte Verläufe mit vitaler Bedrohung geben. Die frühzeitige Erkennung und Behandlung dieser komplikationsträchtigen Situationen stellen die Hauptherausforderung in dieser Krankheitsgruppe dar. Die interdisziplinäre Betreuung ist multimodal, häufig sind mehrere chirurgische Eingriffe erforderlich.
4.3 Konklusion
Bei den strukturellen Anomalien der Orbita sind knöcherne Fehlbildungen der Orbita von okulären Pathologien abzugrenzen. Erstere umfassen vor allem die Kraniosynostosen, welche isoliert und im Rahmen von Syndromen vorkommen können. Okuläre Fehlbildungen können mit orbitalen Läsionen assoziiert sein (z. B. Goldenhar-Syndrom) oder sekundär zu diesen führen. Die Entwicklung der Orbita kann jedoch vollkommen unbeeinflusst von der okulären Organogenese ablaufen. Dermoide sind die häufigsten konnatalen Läsionen der Orbita. Die wesentliche Herausforderung stellen Komplikationen des neurologischen bzw. neurochirurgischen Fachgebietes dar (z. B. Enzephalozelen, Hydrozephalus, etc.).
Zu den erworbenen strukturellen Anomalien gehören in erster Linie postraumatische Veränderungen oder Folgen von Operationen bzw. Bestrahlungen. Diese Veränderungen sind zwar variabel, aber nicht selten.
Die Kenntnis der möglichen strukturellen Veränderungen ist essenziell für eine erfolgreiche Therapie. Die Beachtung der engen Lagebeziehung der Orbita zum Hirnschädel ist hierbei extrem wichtig. Diagnostisch sind entsprechende bildgebende Verfahren zur weiteren topografischen Zuordnung unabdingbar. Für die Therapie ist dabei häufig der interdisziplinäre Austausch mit Neuro- und Ophthalmochirurgen seitens der primär behandelnden Klinik (HNO/Mund-Kiefer-Gesichtschirurgie) zu suchen.
5. Entzündliche Läsionen der Orbita
Es existiert eine Vielzahl unterschiedlicher entzündlicher Läsionen der Orbita. Für eine übersichtliche Darstellung wurde die Einteilung in infektiöse und nicht infektiöse Entzündungen gewählt.
5.1 Infektiöse orbitale Entzündungen
Die infektiöse Zellulitis ist der häufigste Grund für orbitale Entzündungen. Sie entwickeln sich zumeist aus entzündlichen Prozessen der Nasennebenhöhlen, des Gesichts, des Oropharynx sowie der okuläre Adnexstrukturen, zum Teil jedoch auch durch Fremdkörper oder septische Streuung. Die Ursachen umfassen eine große Bandbreite bakterieller und viraler Erreger, Pilze und Parasiten.
5.1.1 Bakterielle Entzündungen (orbitale Zellulitis)
Die häufigsten Ursachen einer infektiösen orbitalen Entzündung sind orbitale Komplikationen einer purulenten Sinusitis, welche in diesem Referat über seltene Erkrankungen der Orbita nicht mit abgehandelt werden. Zur Stadieneinteilung, Pathogenese, Diagnostik und Management möchten die Autoren auf einige wichtige Übersichtsarbeiten zu diesen Krankheitsbildern verweisen [54] [55] [56] [57].
Die Identifizierung seltener Erreger kann eine Herausforderung sein, ist aber für betroffene Patienten von entscheidender Bedeutung, da sie, unabhängig von ihrer Rarität, eine hohe lokale und systemische Morbidität haben können. Opportunistische Infektionen müssen vor allem bei immunsupprimierten und vorgeschädigten Patienten berücksichtigt werden [58]. Der Schlüssel für ein erfolgreiches Management ist ein hohes Maß an Aufmerksamkeit, eine zügige Diagnosestellung sowie die adäquate Therapie der Grunderkrankung.
5.1.1.1 Tuberkulose
Die Häufigkeit der Tuberkulose nimmt in den letzten Jahren durch resistente Stämme und durch die Verbreitung immunsuppressiver Agenzien zu.
-
Pathogenese:
Die Beteiligung der Orbita kann bei der Tuberkulose auf zwei unterschiedlichen Wegen erfolgen [59]:
-
hämatogene Aussaat mit zwei verschiedenen Manifestationsformen in der Orbita:
-
Periostitis (häufigere Form): schleichender Verlauf mit kaltem Abszess, Sequestern und Fisteln; meist junge Patienten betroffen; Lokalisation meist im Jochbein
-
Tuberkulom (seltenere Form): infiltrative orbitale Raumforderung, zum Teil mit neurosensorischem Defizit
-
direkte Ausbreitung aus kontagiösen benachbarten Strukturen (Nasennebenhöhlen oder Glandula lacrimalis): verursacht nekrotisierende, infiltrative Läsionen, z. T. mit kutanen Fisteln [60].
-
Klinik:
-
schmerzhafte Bewegungseinschränkung, keine intraokulären Läsionen, eher unspezifische Symptome, dadurch oft verspätete Diagnose
-
sehr selten auch atypische Mykobakterien (Mycobacterium hominis; Mycobacterium avium)
-
Cave: v. a. bei „kaltem Abszess“, nässenden Höhlen oder (sehr selten) bei durch Optikusneuropathie komplizierter Keilbeinosteomyelitis: klinischer Verdacht zur Diagnosefindung oft wichtiger als positive Blutkultur [58]
-
-
Diagnostik:
-
Therapie:
-
zeitnahe tuberkulostatische Tripeltherapie (Isoniazid, Rifampicin, Etambutol) durch Spezialisten (nicht erst beim Vorliegen einer positiven Kultur, entscheidend ist die passende Konstellation des klinischen und (molekular-)pathologischen Befundes!)
-
Indikation zur chirurgischen Intervention u. a. zur Symptomlinderung, z. B. bei Visusminderung [58]
-
bei Interaktion zwischen HIV und TBC teilweise kompliziertes Management [60]
-
5.1.1.2 Lues
Die durch Treponema pallidum hervorgerufene Geschlechtskrankheit kann gelegentlich (v. a. bei immunsupprimierten und homosexuellen Männern) infolge einer postprimären hämatogenen Aussaat die Orbita befallen und sich dort nach einer Latenzzeit als schmerzhafte Periostitis oder Weichteilläsion (Gummen) manifestieren [61].
-
Klinik:
-
bei posteriorer Manifestation schmerzhaftes Apex-orbitae-Syndrom
-
fokale oder diffuse syphilitische Periostitis in den orbitalen Knochen
-
Auftreten von Weichteilgummen intrakonal, in der extraokulären Muskulatur sowie in der Tränendrüse
-
Diagnostik:
serologische Diagnosesicherung mittels Fluoreszenz-Treponema-Antikörper-Absorptionstest
-
Therapie:
vollständige Abheilung unter antibiotischer Therapie mit Penicillin G [61]
5.1.1.3 Nekrotisierende Fasziitis (NF)
Die nekrotisierende Fasziitis der Orbita ist ein ophthalmologischer Notfall, welcher aufgrund der guten Durchblutung zwar selten auftritt, aber durch sein rasches Fortschreiten bei verzögerter Diagnosestellung zum Tode führen kann [58].

-
Epidemiologie:
Inzidenz von 0,24/100.000 [62]
-
Einteilung [63]:
-
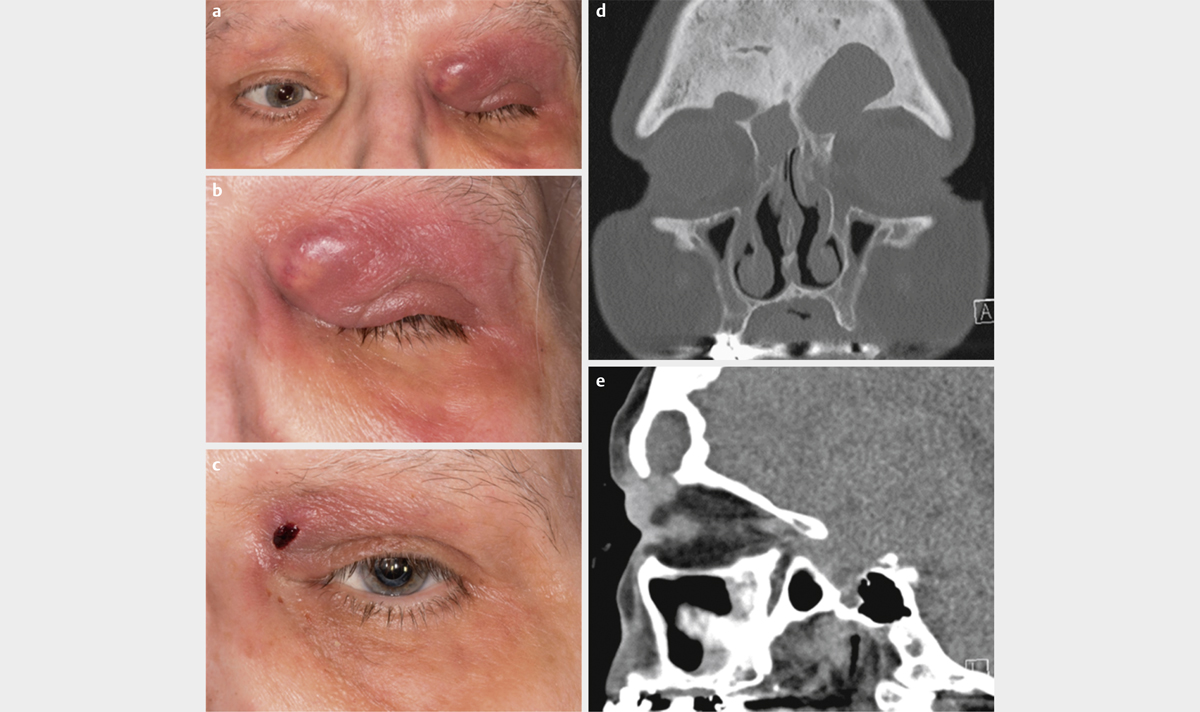
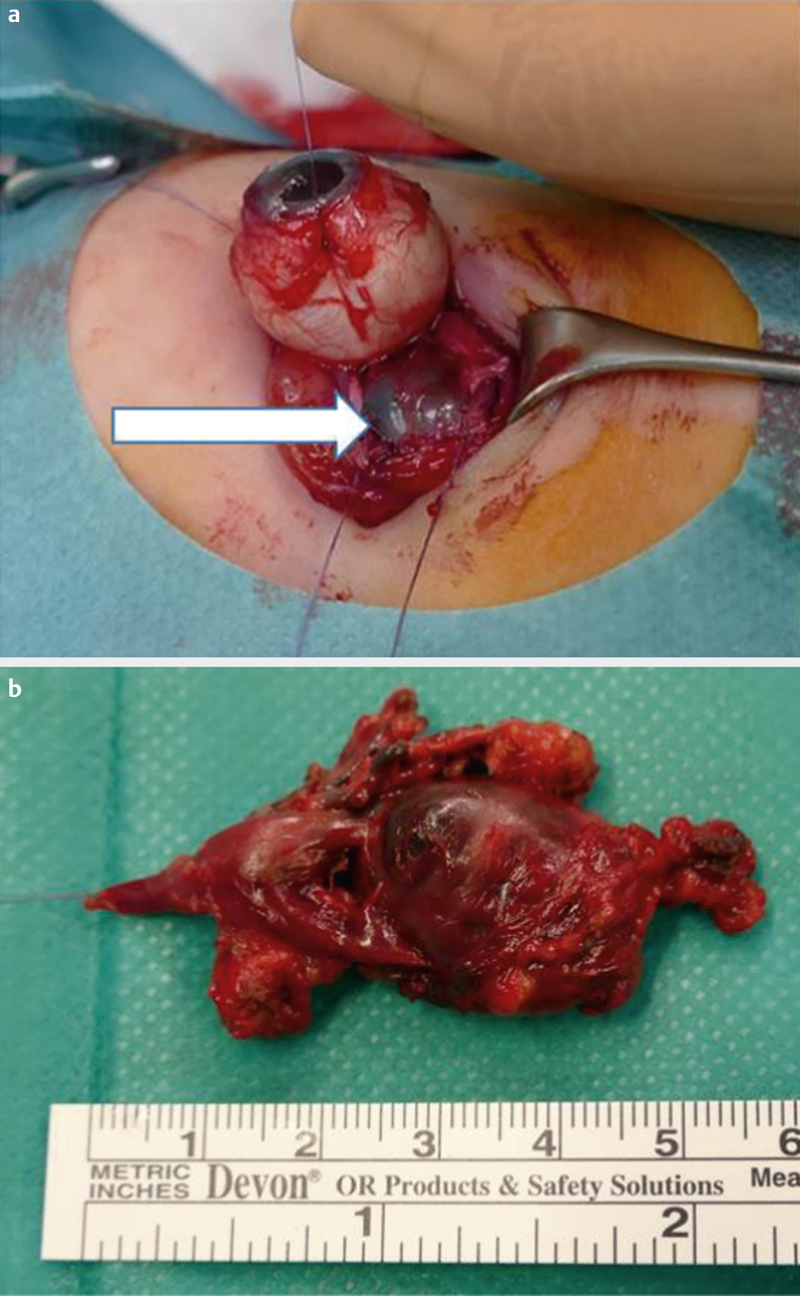
Typ I: polymikrobiell, beispielsweise Aerobier und Anaerobier (v. a. bei multimorbiden Patienten) ([Abb. 7a-d])
-
Typ II: mit ca. 80% häufigster Typ [64] (einzelne bakterielle Erreger, zumeist beta-hämolysierende Streptokokken der Gruppe A (Superantigene und Exotoxine als wichtigste Virulenzfaktoren)
-
Typ III: v. a. Vibriobakterien; in Europa selten vorkommend
-
Typ IV: Pilze
-
-
Ätiologie:
-
Auslöser zumeist penetrierende Traumata (35%), Operationen sowie akute Infektionen der NNH, der ableitenden Tränenwege oder der Haut, außerdem Immunsuppression durch Medikamente oder Malignome
-
in 25% der Fälle Trigger unbekannt [64]
-
Risikofaktoren:
-
hohes Patientenalter, Alkohol- und Drogenabusus, Diabetes mellitus, Kollagenosen und kardiovaskuläre Vorerkrankungen
-
in 50% der Fälle keine Risikofaktoren vorliegend [64]
-
Klinik:
-
aufgrund der dünnen Haut am Auge früh erkennbar
-
noch vor den Hautveränderungen starke Schmerzen möglich („pain out of proportion“) [62]
-
weiterhin Fieber und Schweißausbrüche
-
Entwicklung einer Gangrän innerhalb von Stunden mit livider, blasenbildender Hautverfärbung
-
drohende Erblindung bei Zentralarterienverschluss [58]
-
Toxic-Shock-Syndrom in 30% der Fälle
-
Diagnose:
-
anhand klinischer Symptome
-
Scores, z. B. LRINEC [65], sowie Bildgebung zur schnelleren Diagnose und Abgrenzung gegenüber anderen Infektionen wie Mukormykosen hilfreich
-
Therapie:
-
ausgiebiges Debridement (am Auge hauptsächlich subkutan, zum Teil wiederholt [66] bis hin zur Exenteratio als ultima ratio [67]) in Kombination mit einer Breitbandantibiose (meist Breitspektrumpenicilline oder Cephalosporine plus Clindamycin/Vancomycin; siehe Konsensupapier der dt. HNO-Gesellschaft von 2009 [68])
-
ausschließlich additiver Effekt der hyperbaren Sauerstofftherapie und von Immunglobulinen [63]
-
Prognose:
-
Mortalität schwankend zwischen 8–14 %, insbesondere hoch bei systemischen Komplikationen wie Toxic-Shock-Syndrom mit Multiorganbeteiligung, bei periorbitaler NF mit Erblindung oder Involvierung der Gesichtsweichteile [64] [66]
-
Reduktion der Mortalität durch besondere Aufmerksamkeit und zeitnahe Therapie [58]
5.1.1.4 Nicht infektiöse (abakterielle) Osteomyelitis im Kindesalter
-
Epidemiologie:
-
sehr seltene, nicht infektiöse Entzündung mit mulifokalen knöchernen Läsionen und periodischen Exazerbationen schmerzhafter Schwellungen
-
Synonym: chronische rekurrierende multifokale Osteomyelitis (CRMO)
-
pädiatrisches Äquivalent des SAPHO-Syndroms (Synovitis, Akne, Pustulosis, Hyperostosis, Osteitis)
-
Prävalenz: 1–2/1.000.000
-
Altersgipfel 10. Lebensjahr [69]
-
Ätiologie:
-
multifokale knöcherne Läsionen
-
häufig Assoziation mit anderen chronisch-entzündlichen Erkrankungen, z. B. rheumatoider Arthritis, Morbus Crohn, Sacroiliitis, Psoriasis oder Pyoderma gangraenosum [70]
-
Klinik bei orbitaler Beteiligung:
-
Cephalgien
-
hyperäme Schwellung
-
Diagnostik:
-
CT: osteolytische Auftreibungen
-
Szintigraphie: Focus mit erhöhtem Uptake
-
Histologie: Läsionen mit osteoblastischem/-klastischem Remodeling, granulomatöse Entzündungsinfiltrate mit Riesenzellen
-
Biopsie des beteiligten Knochens zur Absicherung der Diagnose und Ausschluss von Differenzialdiagnosen (z.B. Langerhanszell-Histiozytose, Osteoblastom, Osteosarkom) notwendig
-
Therapie:
-
erste Wahl: NSAID (z. B. Naproxen)
-
alternativ: Bisphosphonate oder Biologika wie TNF-Antagonisten (Etanercept) oder Il-1-Inhibitoren (Anakinra)
-
Prognose:
ungünstiger Verlauf der CRMO möglich (entgegen früheren Annahmen) mit Persistenz von chronischen Schmerzen und physischen Einschränkungen in bis zu 50% der Fälle [71]
-
wichtige Differenzialdiagnose:
infektiöse Osteomyelitis der Orbita
-
ebenfalls seltenes Krankheitsbild im Bereich der Orbita
-
Ätiologie: meist nach vorangegangenen Traumata, Entzündungen (NNH, dentogen oder sekundär infolge einer Bakteriämie bei i.v.-Drogenabusus oder Leukämie) [72]
-
Klinik: bei akutem Verlauf Fieber und Schwellungen, bei chronischen Verläufen Sequester und Fisteln
-
vielfältige Differenzialdiagnosen: von der bakteriellen Zellulitis, atypischen Erregern (Tuberkulose) bis hin zu Neoplasien wie dem Ewing-Sarkom
5.1.2 Orbitale Pilzinfektionen
Pilzinfektionen sind als Ursache für orbitale Entzündungen selten, sollten aber bei immunsupprimierten Patienten immer berücksichtigt werden. Die häufigsten Infektionen erfolgen durch Phytomyzeten (Mucor und Rhizopus spp.) und Ascomyzeten (Aspergillus spp.).
5.1.2.1 Rhino-orbitale Mukormykose
Die Mukormykose ist eine seltene Infektionskrankheit mit hoher Mortalität. Die Diagnose wird häufig verzögert gestellt, die Erkrankung ist durch einen raschen Verfall gekennzeichnet, die zügige chirurgische und medikamentöse Behandlung kann lebensrettend sein [73].
-
Pathogenese:
-
ubiquitär vorkommender Keim, der über Angioinvasion zu Endothelschädigung, Ischämien und dem typischen nekrotischen Schorf (Eschar, fehlt allerdings in frühen Stadien) führt
-
Befall von Lunge, Haut und Gastrointestinaltrakt, am häufigsten aber Nasennebenhöhlen mit Beteiligung der Orbita und letztlich über Orbitaspitze / Lamina cribrosa– auch intrakranieller Befall
-
Ätiologie:
fast immer Assoziation mit einer Grunderkrankung: unkontrollierter Diabetes mellitus (v. a. bei Ketoazidose), Malignome, Verbrennungen, Hämochromatose, Z. n. Nierentransplantation oder Stammzelltransplantation, Einnahme des Chelatbildners Deferoxamin, AIDS
-
Klinik:
-
Allgemeinsymptome: Fieber, Lethargie, Pharyngitis
-
rhinoorbitale Symptome: akute Pansinusitis mit purulenter Rhinorrhoe, Schmerzen und massiver Verborkung (schwarze Verschorfung an Nase und Gaumen), in der Folge Apex-orbitae-Syndrom mit plötzlicher Visusminderung und Ophthalmoplegie, Stupor bei intrakranieller Ausdehnung mit Sinus-cavernosus-Thrombose [58]
-
Diagnostik:
Verdacht auf Mukormykose und rasche bioptische Abklärung bei unklaren Gesichtsbeschwerden, Parästhesien, Schwellungen und rapid-progressiver Sinusitis bei entsprechend vorgeschädigten Patienten [74]
-
Therapie:
-
medikamentöse Behandlung der ersten Wahl: liposomales Amphotericin B
-
weitere medikamentöse Therapie: Isavuconazol und Posaconazol insbesondere zur Salvage-Therapie [73] [75]
-
wiederholtes und ausgedehntes Debridement fast immer notwendig
-
Exenteratio bei Befall Richtung Orbitaspitze erforderlich, bei Befall der vorderen Orbitaabschnitte nicht zwingend notwendig [76]
-
Prognose:
-
früher fast immer infaust, seit Einführung von Amphotericin B Überlebensrate bei 70% [58]
-
in den letzten Jahren Auftreten tödlicher Verläufe auch bei immungesunden Patienten – insbesondere nach Verletzungen oder Insektenstichen in warmen Klimazonen – infolge von Mucorinfektionen durch einen neuen Erregerstamm (Apophysomyces elegans) [58]
5.1.2.2 Aspergillose der Orbita
Aspergillus spp. kommen ubiquitär vor und besiedeln in der Regel als harmlose Saprophyten die Schleimhaut der oberen Atemwege. Als opportunistischer Keim haben sie bei immunsupprimierten Patienten (z. B. nach Nierentransplantation) eine große Bedeutung.

-
Einteilung:
Bei Infektionen im Kopf-Hals-Bereich unterscheidet man zwei Verlaufsformen:
-
non-invasive Form
-
durch eine vorbestehende Läsion der Schleimhaut (meist im Rahmen einer chronischen Sinusitis) Kolonisierung mit Ausbildung sogenannter Fungus Balls oder chronisch-allergische Rhinosinusitis
-
Auftreten auch bei gesunden Patienten
-
Befall der Orbita selten [77]
-
invasive Form
-
bei eingeschränkter Immunabwehr (z. B. durch Leukämie oder AIDS) schnelle Ausbreitung durch Gefäßinvasion mit Gewebsnekrosen, über Ethmoid und Keilbeinhöhle in die Orbitaspitze, den Sinus cavernosus und bis nach intracerebral; Auftreten von Thrombosen und Aneurysmen mit hoher Mortalität [78] [79] [80]
-
hämatogene Streuung aus pulmonalem Infektionsherd oder durch infizierte Nadel bei Heroinabusus möglich[ 61]
-
Klinik:
-
invasive Form: ähnlich fulminantes Bild wie bei der Mukormykose
-
non-invasive Form: meist langsam progrediente, indolente Raumforderung aus den Nasennebenhöhlen mit Ausbreitung Richtung Orbita, häufig vorbestehende Sinusitis
-
isolierter Befall der Keilbeinhöhle mit Optikusneuritis ohne Involvierung der Orbita möglich [81] [82]
-
Diagnostik:
-
Sensitivität und Spezifität der einzelnen Testverfahren zu gering, daher Nachweis durch Kombination aus Bildgebung (CT, MRT), mikrobiologischen Untersuchungen (direkte Mikroskopie, Kultur, PCR, Biomarker) und Histopathologie (astartig septierte Hyphen, chronisch-fibrosierende granulomatöse Entzündung) [83]
-
aufgrund von häufig unspezifischer Symptomatik (geringfügige Sinusitis, retrobulbäre, zum Teil neuropathische Schmerzen, apikale orbitale Entzündung in Bildgebung nur schwer erkennbar) hohes Maß an Aufmerksamkeit bei Diagnosestellung erforderlich [59]
-
wichtige Differenzialdiagnosen:
-
bakterielle Zellulitis, Sinus-Cavernosus-Thombose, Tolosa-Hunt-Syndrom
-
Tumoren der Orbitaspitze oder der Keilbeinhöhle
-
Therapie:
-
antifungale Therapie mit lipsomalem Amphotericin B bis zum Ausschluss einer Mukormykose, dann Umstellung auf das effektivere Voriconazol [84]
-
extensives Debridement des infizierten Gewebes, v. a. bei invasiver Form
-
Prognose:
-
bei immunkompetenten Personen und lokalisierter, nicht invasiver Form bei adäquater Therapie gut [85] ([Abb. 8a–d])
-
bei invasiver Form schlecht
-
bei intrakranieller Ausdehnung immunsupprimierter Patienten fast immer infaust [86].
5.1.2.3 weitere Mykosen
Außerhalb Europas können weitere Erreger für Pilzinfektionen im Bereich der Orbita verantwortlich sein [58] [59]. Diese sind:
-
nordamerikanische Blastomykose
-
Fusariose
-
afrikanische Histoplasmose
-
Sporotrichose
-
Rhinosporidiose
-
Coccidioidomykose
-
Candidiasis
-
Bipolaris hawaiiensis
5.1.3 Virale Infektionen
Orbitale Affektionen durch Viren sind selten, trotzdem können sie durch Infektion oder sekundäre Immunphänomene Krankheiten verursachen. Beispiele dafür sind:
-
Varizella-Zoster-Virus (VZV): akute Myositis, Neuritis Nervus optici
-
Mumpsvirus: akute Dacryoadenitis
-
Epstein-Barr-Virus (EBV): akute Dacryoadenitis, Assoziation mit T-Zell-Lymphomen der Orbita und Nasennebenhöhlen bekannt [61]
5.1.4 Parasitosen
Eine Infestation der Orbita ist auch durch Larven verschiedener Würmer möglich, wobei in Deutschland vor allem die Echinokokkose relevant ist und zu den seltenen Erkrankungen der Orbita gezählt werden kann.
5.1.4.1 zystische Echinokokkose
-
Epidemiologie:
-
weltweit auftretend, v. a. bei engem Zusammenleben von Mensch und Tier
-
meist Patienten zwischen 1. und 4. Lebensdekade [59]
-
endemische Verbreitung in Zentral- und Südamerika, mittlerem Osten, Nordafrika, China und Russland
-
in Deutschland selten (78 Fälle im Jahre 2019 [87])
-
Pathogenese:
-
Ausscheiden von Eiern adulter Bandwürmer aus dem Darm von Carnivoren (z. B. Hunden) mit dem Faeces
-
Aufnahme der Eier durch grasende Herbivoren (z. B. Schafe, Kühe) als Zwischenwirt bzw. vom Menschen selbst durch Verzehr von kontaminiertem Gemüse
-
Verteilung der Eier im Zwischenwirt über den Blutkreislauf ins Gewebe und Aufnahme durch den Endwirt mit der Nahrung
-
Heranwachsen im Endwirt bis zum finalen Larvenstadium
-
Befall von Lunge und Leber am häufigsten, in 1% der Fälle auch Befall der Orbita [88] [89]
-
Klinik:
-
langsam progrediente, indolente, intrakonale Raumforderung
-
intrakranielle Ausbreitung durch Druckarrosion möglich
-
durch Ruptur der Zystenwand (spontan oder im Rahmen der chirurgischen Exzision) fulminante entzündliche oder anaphylaktische Reaktion möglich [61]
-
Diagnostik:
-
sonografisch und im CT gut abgrenzbare zystische Struktur mit KM-aufnehmendem Randsaum [90]
-
Hämagglutinations- und ELISA-AK-Test möglich, bei isoliertem orbitalem Befall aber nur zu 50% sensitiv [61]
-
Therapie:
-
alleinige Therapie mit Albendazol für 3 Monate, bei Begleitentzündung zusätzlich systemisch Kortikosteroide
-
Kombination aus Albendazol, Praziquantel und chirurgischer Exzision
-
bei unklarer Diagnose oder kompressionsbedingter Visusminderung chirurgische Exzision notwendig [61], Feinnadelaspiration sowohl diagnostisch als auch therapeutisch (Albendazolinjektion) einsetzbar [59]
5.1.4.2 Zystizerkose
-
Epidemiologie:
endemisches Vorkommen in Ländern mit schlechten sanitären Bedingungen (Lateinamerika, Teile von Asien sowie südlich der Sahara)
-
Pathogenese:
-
durch den Parasiten Taenia solium (Schweinebandwurm) hervorgerufen
-
Aufnahme der Eier durch ungares Schweinefleisch und hämatogene Verteilung
-
Ausbilden zystisch abgekapselter Larven mit bevorzugter Ansiedlung in Auge und Gehirn (Neurozystizerkose, lebensbedrohlich) [61]
-
Klinik:
-
am Auge oft subkonjunktivaler, intravitrealer und retinaler Befall
-
im Bereich der Orbita vor allem extraokuläre Muskulatur betroffen [93] [94]
-
Therapie:
-
chirurgische Exzision und medikamentöse Therapie mit Albendazol oder Praziquantel
-
Reduktion der Begleitentzündung durch Steroide [95]
5.1.4.3 Trichinose
-
Pathogenese:
-
Aufnahme der abgekapselten Larven durch Ingestion von ungarem Schweinefleisch
-
im Darm Entwicklung der Larven zu Rundwürmern, Ausbildung neuer Larven und hämatogene Streuung
-
Ausbildung von Zysten in der quergestreiften Muskulatur
-
Klinik:
-
Erstmanifestation häufig in der extraokulären Muskulatur
-
präseptales Ödem, Chemosis und schmerzhafte Myopathie
-
Therapie:
medikamentös mit Thiabendazol [59] [61]
5.1.4.4 Ophthalmomyiasis
-
Ätologie:
durch Larven der Dasselfliege in tropischen Regionen hervorgerufen
-
Klinik:
Symptome variieren von leichter Konjunktivitis bis hin zu schwerer Destruktion der gesamten Orbita
-
Therapie:
-
Entfernung der Larven
-
Desinfektion bis hin zur Exenteratio bei schwerem Befall
5.1.4.5 Weitere Parasitosen und Fliegenlarven mit möglicher Augenbeteiligung [96]
-
Cestoden
-
Dirofilarien
-
Loa Loa
-
Onchocerva volv.
-
Oxyuren
-
Plasmodien
-
Porocephalus armillatus
-
Schistosoma
-
Thelazia
-
Wuchereria bancrofti
5.2 Nicht infektiöse orbitale Entzündungen
5.2.1 Einleitung
Orbitale Entzündungen stellen streng genommen keine Diagnose im engeren Sinne dar [97]. Die Entzündung entspricht der adaptiven Antwort des Immunsystems auf eine erkennbare Ursache, beispielsweise auf eine Infektion oder Verletzung, aber auch auf Autoimmunerkrankungen. Bei 20% der raumfordernden Läsionen der Orbita besteht eine Assoziation mit einer Entzündung [98]. Das Auffinden der ursächlichen Erkrankung stellt dabei oft eine große Herausforderung dar. Gemeinsame Merkmale sind die klinischen Entzündungszeichen sowie das histologische Bild einer polymorphen Entzündungszellinfiltration [59]. Bezüglich des Auftretens sowie der Intensität besteht eine ausgeprägte Variabilität [99]. Auf die endokrine Orbitopathie als häufigste nicht infektiöse entzündliche Veränderung der Orbita soll in diesem Referat über seltene Erkrankungen nicht näher eingegangen werden.
5.2.2 Idiopathische Orbitaentzündung (IOE)
Die idiopathische Orbitaentzündung, früher unter dem Begriff „Pseudotumor orbitae“ bekannt, umfasst eine heterogene Gruppe von Erkrankungen, die durch eine orbitale Inflammation ohne identifizierbare lokale oder systemische Ursache gekennzeichnet sind. Es handelt sich dabei um eine seltene Entität sowie um eine Ausschlussdiagnose [100]. Sie ist charakterisiert durch eine Größenzunahme oder Raumforderung orbitaler Strukturen, welche durch unspezifische entzündliche, zum Teil fibrosierende, selten auch destruktive Läsionen hervorgerufen werden kann [61]. Die Pathophysiologie ist nicht vollständig geklärt, diskutiert wird eine autoimmune Genese, welche durch Infektionen getriggert werden kann [99]. Es wurden diverse Einteilungen vorgeschlagen, aufgrund der sehr variablen klinischen und pathologischen Merkmale hat sich bislang keine davon durchsetzen können [101] [102] [103].
Die IOE kann jede Struktur der Orbita befallen, die Erstmanifestation variiert von einem akuten Beginn bis zu einem schleichenden Verlauf.
Nach der Lokalisation werden unterschieden [61]:
-
idiopathische Dakryoadenitis: isolierter Befall der Tränendrüse (häufigste Variante)
-
idiopathische orbitale Myositis: begrenzt auf einen oder mehrere extraokuläre Muskeln
-
diffuse IOE mit Befall verschiedener orbitaler Strukturen (seltener)
-
idiopathische Perineuritis des Nervus opticus (sehr selten): Befall der Optikusnervenscheide
-
anteriore IOE: von einigen Autoren als eigenständige Entität aufgeführt [103]
Die Liste der Differenzialdiagnosen ist lang und umfasst nahezu alle Formen infektiöser und nicht infektiöser orbitaler Entzündungen sowie eine Vielzahl neoplastischer sowie vaskulärer Läsionen [99]. Die Diagnose kann nur histologisch gestellt werden, sodass Biopsien eine wichtige Rolle spielen, insbesondere bei fortgeschrittenen Fällen und Rezidiven, um relevante Differenzialdiagnosen auszuschließen. Ausnahmen sind die idiopathische Myositis mit ihrem charakteristischen klinischen und radiologischen Bild sowie Läsionen im Bereich der Orbitaspitze, wo die Risiken durch iatrogene Verletzungen höher sein können als der erwartete Benefit. Ein gutes Ansprechen auf Kortikosteroide findet man bei einer Vielzahl unterschiedlicher orbitaler Läsionen inklusive der endokrinen Orbitopathie und der Malignome, sodass dies allein nicht zur Diagnosestellung genutzt werden sollte.
-
klassischer orbitaler Pseudotumor:
-
breites inflammatorisches Substrat von lymphozytären, granulozytären und gelegentlich histiozytären Infiltraten, organisiert in Lymphfollikeln mit Keimzentren
-
erhöhter Bindegewebsanteil mit Ödem und Fibrose
-
frühzeitig erkennbare Zerstörung der Tränendrüsen-Azini bzw. der Muskelfasern
-
Abgrenzung zu lymphoproliferativen Erkrankungen wichtig
-
idiopathisch sklerosierende Form:
-
Gewebssklerose und Hyalinisierung dominierend
-
Mangel an entzündlichem Infiltrat, Ähnlichkeiten mit retroperitonealer Fibrose
-
Ausrichtung der Diagnostik auf IgG4-assoziierte Erkrankung [104]
-
Seltener granulomatöse (histiozytäre Riesenzellinfiltration) und vaskulitische (Vaskulitis der kleinen Gefäße) Entzündungsreaktionen
-
Übergänge vom klassischen orbitalen Pseudotumor zum sklerosierenden Subtyp im Verlauf möglich [100]
-
Klinik:
-
sehr variabel, typischerweise unilateral mit einer Anamnese von Tagen bis Wochen, von leichten Anzeichen einer Entzündung bis zu schwersten Schmerzen und orbitalen Drucksteigerungen mit der Gefahr der Optikuskompression, häufig Rezidive beim klassischen Pseudotumor sowie der sklerosierenden Form [100]
-
je nach Lokalisation schmerzhafte (schwellungsbedingte) Pseudoptosis, Chemosis, Exophthalmus sowie (meist schmerzbedingt) eingeschränkte Bulbusmotilität
-
sklerosierende Form: häufig Schmerzen, eher dezente Lidschwellung, okuläre Bewegungseinschränkung, Visusminderung bis zum Visusverlust ([Abb. 9a, b])
bildgebende und serologische Diagnostik:

-
idiopathische Dakryoadenitis / idiopathische Myositis: Vergrößerung der betroffenen Strukturen bei unveränderter Differenzierung
-
sklerosierende Form: homogen anreichernde, unscharf begrenzte Infiltrate verschiedener orbitaler Strukturen (initial zumeist lateral mit Befall der Tränendrüse), im weiteren Verlauf Ausbreitung Richtung Sinus cavernosus und Fossa pterygopalatina möglich, außerdem evtl. multifokale Fibrosklerose (insbesondere retroperitoneal) ([Abb. 9c, d])
-
breite laborchemische und serologische Diagnostik zur differenzialdiagnostischen Abgrenzung und Einschätzung der Entzündungsaktivität sinnvoll (u. a. Nieren- und Schilddrüsenwerte, ANCA, ACE, Rheumafaktoren und IgG4-Serumspiegel)
-
Therapie:
-
bei hohem Leidensdruck der Patienten meist zeitnahe Therapie erforderlich
-
verschiedene Stufen der antiinflammatorischen Therapie von alleiniger Observatio über Antibiotika, NSAID, Steroide bis hin zu weiteren Immunsuppressiva [100]
-
zumeist gutes Ansprechen auf eine systemische Hochdosistherapie mit Kortikosteroiden, bei der sklerosierenden Form schlechtes Ansprechen auf Kortikosteroide und Bestrahlung, durch rasche Diagnosestellung und schnellen Beginn einer Kombinationstherapie aus Kortikosteroiden und Immunsuppressiva (gemeinsam mit Rheumatologen) zumindest Verzögerung der Progression auch beim sklerosierenden Subtyp möglich [59]
-
bei therapierefraktären und chronischen Verläufen systemische Gabe von Immunsuppressiva (z. B. Methotrexat und Azathioprin), Biologika wie Rituximab sowie eine Kombination mit Bestrahlung der Orbita
-
Chirurgie als Debulking bei der lacrimalen Form, Dekompression der Orbita bei Kompression des Nervus opticus, Exenteratio orbitae als ultima ratio bei persistierenden massiven Schmerzen [99]
5.2.2.1 Tolosa-Hunt-Syndrom
-
Epidemiologie:
-
seltene idiopathische granulomatöse Entzündung des Sinus cavernosus und der Orbitaspitze mit Kompression der benachbarten Hirnnerven (CN III – CN VI)
-
jährliche Inzidenz von 1/1.000.000 [105]
-
Klinik:
-
einseitige Ophthalmoplegie und (z. T. migrierende) Paresen der entlang des Sinus cavernosus und der Fissura orbitalis superior verlaufenden Hirnnerven (Nervus oculomotorius am häufigsten betroffen)
-
häufig assoziiert mit konstanten retrobulbären Gesichtsschmerzen
-
Diagnose:
Diagnosesicherung mittels MRT oder Biopsie, um Differenzialdiagnosen (Sarkoidose, Malignome, Sinus-cavernosus-Thrombose, Karotis-Cavernosus-Fistel) auszuschließen [106]
-
Therapie:
-
initial Hochdosis-Glukokortikoid-Stoßtherapie, dann mindestens über 4–6 Wochen ausschleichen (ggf. auch länger)
-
weitere Optionen: Radiotherapie und Immunsuppressiva (z. B. Infliximab) [107] [108]
-
Prognose:
-
rasche Linderung der Schmerzen durch Kortikosteroide
-
Cave: auch bei Malignomen kann es nach hochdosierter Anwendung von Kortikosteroiden zu temporärer Beschwerdebesserung kommen, siehe Fallbeispiel zu [Abb. 10a–f].
-
Regredienz der Neuropathien häufig erst nach Monaten, z.T. persistierend

Fallbeispiel 1
-
Anamnese:
-
79-jähriger männlicher Patient mit seit Monaten progredienten, periorbitalen Kopfschmerzen rechts: intermittierend, neuralgischer Charakter im Bereich Nervus ophthalmicus, Berührungsempfindlichkeit
-
seit Wochen Trochlearis- und Abduzensparese rechts (Doppelbilder)
-
Diagnostik:
-
MRT der Orbita: KM-anreichernde Struktur im Extrakonalraum am Orbitadach (a. e. verdickter Nervus supraorbitalis), bis in die Fissura orbitalis superior zu verfolgen, fokale KM-Anreicherung im Orbitatrichter bis in den Sinus cavernosus ([Abb. 10a–c])
-
PET-CT: keine malignomtypische Glukosekonsumption, insbesondere im Bereich der Orbita
-
Lumbalpunktion: kein Hinweis auf eine floride Entzündungsreaktion des ZNS, keine autochtone Immunglobulinsynthese, keine oligoklonalen Banden, serologisch keine Hinweise auf eine akute Infektion mit neurotropen Erregern.
-
Weiterer Verlauf:
-
Arbeitsdiagnose: V. a. Tolosa-Hunt-Syndrom. Differenzialdiagnostische Überlegungen gehen in Richtung einer infektiösen Genese, systemisch-granulomatösen Entzündung oder Neoplasie.
-
Interdisziplinäre Fallberatung: Therapieversuch mit systemischer Glukokortikoidgabe (auch wegen nicht unerheblichem periprozeduralem Risiko im Falle einer Biopsie) und Analgetika
-
zunächst Beschwerdebesserung, (v.a. der Bulbusmotilität) und regredienter MRT-Befund, DD autoimmune Genese, Malignom nicht sicher auszuschließen
-
Fortführung der Glukokortikoidtherapie
-
bei vorgeschädigter Lunge (Fibrose) unter immunsuppressiver Therapie wiederholt Pneumonie bzw. Influenza-Pneumonitis
-
nach 3 Monaten trotz regredientem MRT-Befund Indikation zur Biopsie wegen persistierender Beschwerden histologische Diagnose: schlecht differenziertes Adenokarzinom weitere Stagingdiagnostik ohne wegweisende Befunde, somit a.e. Primärtumorlokalisation
-
Therapie:
Strahlentherapie (unter Berücksichtigung von Komorbidität und Patientenwunsch)
Auch bei entzündlich imponierenden Prozessen der Orbita muss ein malignes Geschehen in Betracht gezogen werden. Eine regrediente Beschwerdesymptomatik unter Glukokortikoidtherapie schließt eine maligne Erkrankung nicht aus.
5.2.2.2 IgG4-assoziierte Orbitopathie

-
Definition:
subakut bis chronisch verlaufende immunologische Systemerkrankung mit derben lymphozytären Infiltraten und fibrotischen Läsionen [111]
-
Lokalisationen und Klinik:
-
häufigste Manifestationen sind die autoimmune Pankreatitis, sklerosierende Cholangitis, Nephritis und renale Fibrose, Befall von Tränendrüsen, Speicheldrüsen und Lymphknoten [111]
-
Manifestationen im Bereich der Orbita typisch, nahezu jede Struktur kann betroffen sein, insbesondere jedoch
-
Tränendrüse: „IgG4-assoziierte Dacryoadenitis“ mit meist schmerzloser Schwellung, meist bilaterale Ausprägung und mit Sicca-Symptomatik [112]
-
orbitale Nerven und Muskulatur: vorrangig Nervus infraorbitalis; meist schmerzlos, mit Diplopie und vergrößerter Tränendrüse
-
orbitales Fettgewebe: mit vergrößerten Tränendrüsen einhergehend [112] [113]
-
selten auch destruierende Läsion mit Enophthalmus [113] [114], Befall okulärer Strukturen oder der knöchernen Begrenzung
-
bei mehr als 70% der Patienten auch extraokuläre Manifestationen [111]
-
Sehstörungen als führendes Symptom [111]
-
Diagnostik:
-
Bildgebung:
-
diffus vergrößerte Tränendrüse, Muskulatur (vor allem lateral), Nervus infraorbitalis, z. T. mit Auftreibung des knöchernen Kanals ([Abb. 11a, b])
-
häufig ipsilaterale Affektion der Nasennebenhöhlen [112]
-
Histologie:
-
lymphoplasmazelluläre Infiltration mit obliterierender Phlebitis und eosinophiler Entzündung
-
Anteil an IgG4-positiven Plasmazellen > 40 % (auslösender Stimulus für Differenzierung von B-Lymphozyten in IgG4-produzierende Plasmazellen bislang unklar)
-
Fibrosierung im weiteren Verlauf
-
histologische Diagnosekriterien: IgG4/IgG-Ratio > 50% sowie > 30 IgG4-positive Zellen pro Hochvergrößerungsfeld (High Power field, HPF) [115]
-
Labor:
-
erhöhter IgG4-Serum-Spiegel, allerdings unspezifisch (Erhöhung auch bei Churg-Strauss-Syndrom, Sarkoidose, allergischen Erkrankungen) und insbesondere bei limitiertem orbitalen Befall nicht nachweisbar
-
ansonsten keine systemischen Entzündungszeichen [99]
-
Therapie:
-
erste Wahl: bisher orale Glukokortikoide (30–40 mg/d) als Induktionstherapie, niedrig-dosierte Erhaltungstherapie über Jahre, hierunter häufig Beschwerderegredienz
-
Empfehlung aus neueren Studien: Einsatz von Rituximab nicht nur bei Rezidiven, sondern auch in der Erstlinientherapie; darunter deutlich bessere Erfolgsraten als sonstige disease modifying anti rheumatic drugs (DMARD) wie Methotrexat, Mykophenolat und Azathioprin [116]
-
chirurgisches Debulking und Strahlentherapie nur noch selten indiziert [116] [117]
Fallbeispiel 2
-
Anamnese:
-
75-jähriger Patient, Z. n. funktioneller endonasaler endoskopischer Nasennebenhöhlenoperation (FESS) 2007
-
vor sechs Monaten Augenschwellung rechts und Doppelbilder seit 6 Monaten, auswärts Re-FESS bei klinischem Befund eines „Orbitaabszesses“
-
drei Monate später Re-FESS bei polypöser Pansinusitis mit orbitaler Beteiligung ebenfalls auswärtig
-
bei Erstvorstellung persistierende Doppelbilder, Visusminderung, Lidödem, Sensibilitätsstörungen
-
Diagnostik:
-
MRT: extrakonale Raumforderung am Orbitaboden, die nicht sicher vom Musculus rectus inferior zu trennen ist und Kontrastmittel anreichert ( [Abb. 11a, b] )
-
nach Biopsie über transkonjunktivalen Zugang Diagnose einer IgG4-assoziierten orbitalen Entzündung
-
Therapie (gemeinsam mit Rheumatologie):
-
Glukokortikoide und Methotrexat, darunter dauerhafte Beschwerdefreiheit
5.2.3 Vaskulitiden im Bereich der Orbita
Vaskulitiden haben unter den orbitalen Entzündungen eine besondere Bedeutung. Sie sind häufig Teil einer Systemerkrankung und initial nicht immer erkennbar, haben für den Patienten aber mitunter weitreichende Konsequenzen. Histopathologisches Hauptmerkmal sind angiodestruktive Prozesse mit deutlicher Begleitentzündung. Die Vaskulitiden umfassen eine Vielzahl von Erkrankungen. Für die Orbita sind vor allem die orbitale Granulomatose mit Polyangiitis (GPA, früher Morbus Wegener), die allergische Granulomatose (Churg-Strauss-Vaskulitis) sowie die Polyarteriitis nodosa von Bedeutung, weitere Vaskulitiden sind mit ihren Hauptmerkmalen kurz aufgeführt.
5.2.3.1 Vaskulitiden der kleinen und mittleren Gefäße
5.2.3.1.1 Orbitale Granulomatose mit Polyangiitis (GPA)
-
Definition:
nekrotisierende granulomatöse Entzündung des Respirationstraktes und nekrotisierende Vaskulitis kleiner und mittlerer Gefäße der Atemwege und der Niere
-
Epidemiologie:
-
seltene Erkrankung, Inzidenz 3/100.000 [118]
-
Beteiligung der Orbita in ca. 50% aller Fälle [111] (nahezu jede Struktur kann betroffen sein)
-
Einteilung in zwei klinische Verlaufsformen [119]:
-
limitierte GPA:
-
zumeist Befall der oberen Atemwege
-
meist kein Nachweis von ANCA
-
generalisierte GPA:
-
Befall von Atemwegen, Nieren, weiteren Organen
-
häufig Assosziation mit c-ANCA- (90%) und p-ANCA-Erhöhung (20%) [111]
-
früher hohe Mortalität, durch frühzeitige Kombinationstherapie aktuell bis zu 90% Remissionen [120]
-
Symptomatik [111]:
-
Proptosis als Schlüsselmerkmal (im Zusammenhang mit pulmonalen und renalen Symptomen wegweisend für korrekte Diagnosestellung)
-
schmerzhafte Ophthalmoplegie durch Ausbreitung der orbitalen Raumforderung in benachbarte Strukturen (z. B. Sinus cavernosus)
-
Epiphora durch Obstruktion der ableitenden Tränenwege bei NNH-Befall, Diplopie durch Vaskulitis der Gefäße der extraokulären Muskulatur (EOM), Inflammation der Glandula lacrimalis, Bulbusperforation durch nekrotisierende Skleritis, Xanthelasmen-ähnliche Verfärbung der Augenlider („yellow lid sign“)
-
visusbedrohende Kompression des Nervus opticus bei Infiltration benachbarter Strukturen
-
meist subakuter Beginn, meist bilateraler Befall im Verlauf, Episoden mit deutlicher Befundprogression im Rahmen des chronischen Erkrankungsverlaufs typisch
-
im Verlauf erhebliche Destruktion durch Fibrosierung und orbitale Atrophie mit Enophthalmus, Motilitätsstörungen und chronischen Schmerzen möglich [118] [121]
-
Diagnostik:
-
Bildgebung (CT und MRT) [122]
-
extrakonale Infiltrate, vorrangig inferomedial in enger Nachbarschaft zu den betroffenen Nasennebenhöhlen mit knöcherner Destruktion und Septumperforation
-
isolierter Befall der Orbitaspitze möglich (z. T. mit intrakranieller Ausbreitung) oder isolierter Befall der Glandula lacrimalis (diffuse Vergrößerung der Drüse)
-
diffuse Ausbreitung intra- und extrakonal („wall-to-wall“-Tumor) als pathognomonisches Zeichen für fortgeschrittenen Befall
-
MRT zur Identifizierung einer granulomatösen Entzündungsreaktion geeignet
-
Pathologie:
-
klassische histologische Trias aus Vaskulitis, Nekrose und granulomatöser Entzündung in der Orbita nur bei 50% der Patienten
-
weitere Merkmale: polymorphe entzündliche Infiltrate, degenerativ veränderte Granula/Kollagen sowie in vielen Fällen erhöhte Anzahl IgG4-positiver Plasmazellen [123]
-
auch bei ANCA-negativer GPA: immunhistochemische Zusatzuntersuchungen auf IL-17 und IL-23 für Differenzierung zur Sarkoidose [111]
-
Therapie:
-
Management der orbitalen GPA analog zur systemischen GPA
-
aktueller Standard: Kombination von Kortikosteroiden mit Cyclophoshamid, Methotrexat oder Azathioprin; durch frühzeitigen Therapiebeginn Vermeidung fataler lokaler oder systemischer Verläufe [118]
-
bei therapierefraktären Fällen: Rituximab [124] (besseres Nebenwirkungsprofil bei vergleichbarer Wirksamkeit)
-
chirurgisches Debulking bei persistierender Proptosis, unkontrollierbaren Schmerzen oder kompressiver Neuropathie des Nervus opticus [121]
5.2.3.1.2 Allergische Granulomatose (Churg-Strauss-Syndrom)
-
Definition:
-
allergische Systemerkrankung; initial Manifestation mit Asthma und Rhinosinusitis, im Verlauf nekrotisierende Vaskulitis kleiner und mittlerer Gefäße und eosinophile granulomatöse Entzündung [118]
-
orbitaler Befall sehr selten
-
Lokalisation:
vorrangiger Befall von Lunge, Nasennebenhöhlen (NNH) und peripherem Nervensystem
-
Klinik:
-
zum einen als ischämische Vaskulitis, zum anderen als orbitaler entzündlicher Pseudotumor mit Dacryoadentis, Myositis, Periskleritis, Perineuritis
-
kann Erstmanifestation des Syndroms sein [125] ([Abb. 12a–c])
-
im Verlauf Proptosis, Diplopie, Ophthalmoplegie, Visusminderung durch Optikuskompression
-
im vaskulitischen Stadium lebensbedrohliche Komplikationen möglich (Peritonitis durch Darmperforation, eosinophile Myokarditis mit Myokardinfarkt)

Diagnostik:
-
Biopsie aus den betroffenen Strukturen
-
histologisches Hauptmerkmal: nekrotisierende Vaskulitis mit extravaskulären eosinophilen Infiltraten ([Abb. 12d])
-
serologisch positive p-ANCA in 70% der Fälle [126]
-
Bildgebung (CT, MRT): unspezifische Veränderungen mit Größenzunahme des Retrobulbärraums, Schwellung von Tränendrüse und extraokulärer Muskulatur (EOM)
-
Diagnosekriterien des American College of Rheumatology (1990; 4 von 6 müssen zutreffen): Asthma, Abnormalitäten der Nasennebenhöhlen, pulmonale Infiltrate, Bluteosinophilie (> 10% der Leukozyten), Gewebseosinophilie, Mono-/Polyneuropathie [127]
-
Therapie:
-
Erstlinientherapie: systemische Kortikosteroide (oral, intravenös), Methotrexat und Cyclophosphamid additiv als Steroidsparer bei kritischer Organbeteiligung
-
Biologika bei Rezidiven oder refraktären Fällen: Rituximab, Infliximab, Mepolizumab oder Omalizumab [118] [128], in der Regel medikamentöse Dauertherapie notwenig
-
Prognose:
vor der Ära der Biologika: Mortalität von bis zu 50% bei Befall von zwei oder mehr Organen [129]
5.2.3.1.3 Polyarteriitis nodosa
-
Definition:
-
nekrotisierende Vaskulitis der mittleren und kleinen Arterien sowie gelegentlich der Begleitvenen
-
segmentaler Befall charakteristisch (Bifurkationen bevorzugt)
-
typischerweise Thrombosen und noduläre Aneurysmen [59]
-
histologisches Merkmal: fibrinoide Nekrose der Media sowie das gleichzeitige Vorliegen mehrerer Stadien der Entzündung
-
Epidemiologie:
-
meist männliche Patienten in der 4. bis 5. Lebensdekade
-
Assoziation zu Hepatitis B
-
Orbita bei ca. 10% der Patienten befallen [118], gelegentlich Erstmanifestation der Erkrankung
-
Lokalisation:
Prädilektionsstellen: Niere, Herz, Leber, Gastrointestinaltrakt, periphere Nerven und ZNS, aber auch Knochen und Gelenke
-
Klinik:
-
typische ophthalmologische Läsionen: retinale Vaskulitis, choroidale Infarkte mit Exsudationen, ischämische Optikusneuropathie
-
Sekundärphänomene der Vaskulitis: Ischämie der extraokulären Muskulatur (EOM) mit Ophthalmoplegie, diffuse orbitale Inflammation mit Exophthalmus, Motilitätseinschränkung, Chemosis
-
Diagnosestellung anhand der Kombination aus orbitaler Entzündung mit anderen Symptomen der Erkrankung, unspezifischen Entzündungszeichen im Labor, Biopsie und Bildgebung (ggf. angiografischer NAchweis von Aneurysmen)
-
Therapie:
-
systemische Kortikosteroide und Cyclophosphamid
-
Einsatz anderer DMARD wie Methotrexat, Azathioprin und Rituximab ebenfalls beschrieben [118]
-
Prognose:
5-Jahres-Überlebensrate bei 80% [118]
5.2.3.2 Vaskulitiden der großen Gefäße
5.2.3.2.1 Riesenzellarteriitis (Arteriitis temporalis)
-
Definition:
-
idiopathische Vaskulitis mittlerer und großer Arterien der Kopf-Hals-Region
-
histologisch segmentale Entzündung mit Riesenzellen, Lymphozyten, Plasmazellen und Eosinophilen [118]
-
Epidemiologie:
-
Altersgipfel 7. bis 8. Lebensdekade
-
meist Frauen betroffen [111]
-
orbitale Beteiligung selten
-
Ätiologie:
Kombination aus genetischer Prädisposition (kaukasische Rasse) und Umweltfaktoren (Assoziation mit Virusinfektionen, z. B. VZV, CMV, Parvovirus 19) [130]
-
Klinik:
-
Allgemeinsymptome:
-
Kopfschmerzen, Schmerzen beim Kauen („jaw claudication“)
-
B-Symptome wie Fieber, Müdigkeit, Gewichtsverlust
-
Polymyalgia rheumatica, Empfindlichkeit der Kopfhaut [111]
-
ophthalmologische Symptome:
-
Visusminderung durch Ischämie des Nervus opticus (anteriore ischämische Optikusneuropathie [A-AION]: dringliche Behandlungsindikation)
-
weiterhin Diplopie, chorioidale und retinale Ischämiezeichen [111]
-
orbitale Symptome:
Ophthalmoplegie und Diplopie (durch Ischämie und Inflammation) [131]
-
Diagnostik:
Diagnosestellung durch Kombination aus typischen Symptomen (Kopfschmerzen, Empfindlichkeit der Kopfhaut), Laborparametern (erhöhte BSG), radiologischen Befunden (KM-Enhancement der Orbita im MRT) und Biopsie der Arteria temporalis (Biopsie der orbitalen Läsion selten notwendig) [118]
-
Therapie:
-
First-Line-Therapie: Hochdosis-Kortikosteroide (Cave: Langzeitnebenwirkungen)
-
aktuelle Studienergebnisse: Tocilizumab (Anti-IL-6) vielversprechend [132]
5.2.3.2.2 Takayasu-Arteriitis
-
Definition:
granulomatöse Panarteriitis mit Affektion der Aorta und deren Hauptästen
-
Epidemiologie:
-
Erstbeschreibung bei japanischen Frauen mittleren Alters
-
aktuelle Untersuchungen: alle ethnischen Gruppen betroffen, Prävalenz zunehmend [133]
-
Klinik:
-
vaskulitische Destruktion der Arteria carotis mit kollateraler Blutversorgung zum Auge
-
Visusverlust durch ischämische okuläre Komplikationen möglich
-
initial Amaurosis fugax oder progressiver permanenter Visusverlust möglich [134]
-
Diagnostik:
-
Fluoreszenz-Angiografie zum Nachweis einer kritischen Perfusion der Retina
-
in den letzten Jahren ersetzt durch MR-Angiografie und FDG-PET
-
Therapie:
-
primär Glukokortikoide
-
bei steroidrefraktären Fällen: Leflunomid und Tocilizumab [135]
5.2.3.3 Vaskulitiden von Gefäßen variabler Größe
5.2.3.3.1 Cogan’s Syndrom
-
Definition:
-
seltene entzündliche Systemerkrankung
-
Hauptbeteiligung des visuellen und audiovestibulären Systems sowie von Gefäßen variabler Größe [111]
-
Epidemiologie:
-
meist junge Erwachsene betroffen
-
Altersgipfel 29 Jahre
-
bisher ca. 250 Fälle publiziert [136]
-
orbitale Beteiligung extrem selten
-
-
Ätiologie und Pathogenese:
-
autoimmune Genese mit Ursprung im Innenohr
-
Triggerung durch initiale Virusinfektion
-
Identifikation verschiedener Autoantigene (CD 148, Connexin 26) [137]
-
Einteilung:
-
typische Form:
non-infektiöse interstitielle Keratitis mit Menière-ähnlicher vestibulochleärer Funktionsstörung innerhalb von zwei Jahren nach Erstmanifestation [111]
-
atypische Form:
-
verschiedene okuläre Symptome wie Konjunktivitis, Episkleritis, Glaukom, Uveitis; Labyrinthaffektion zwei Jahre nach Erstmanifestation
-
Hörverlust teilweise sehr ausgeprägt, beidseitige Taubheit bei 60% der Patienten [138]
-
Klinik:
-
durch nekrotisierende Vaskulitis in ca. 70% der Fälle Systembeteiligung mit Manifestation an Blutgefäßen (Aorta, Nieren- und Koronargefäße), neurologischen und gastrointestinalen Symptomen; schwerer, z. T. lebensbedrohlicher Verlauf möglich
-
bei orbitaler Beteiligung (nur Fallberichte): Hörverlust, Vertigo, Enophthalmus und orbitale Inflammation (jedoch ohne bioptischen Nachweis der Genese) [118]
-
Diagnostik:
neurootologische, ophthalmologische und angiologische Diangostik zum Ausschluss von Differenzialdiagnosen
-
Differenzialdiagnosen:
-
kongenitale Syphilis, Susac-Syndrom, Vogt-Koyanagi-Harada-Syndrom
-
sonstige Vaskulitiden, z. B. Granulomatose mit Polyangiitis [136]
-
Therapie:
-
First-Line-Therapie: systemische Hochdosis-Glukokortikoide, hierdurch häufig Verbesserung der vestibulocochleären und okulären Symptomatik [139]
-
Rezidivprophylaxe: Biologika wie Infliximab (Einsatz auch als First-Line-Medikament gemeinsam mit Glukokortikoiden zu einem frühen Zeitpunkt der Erkrankung zur Vermeidung irreversibler Schäden propagiert) [140]
5.2.3.3.2 Morbus Behçet
-
Definition:
idiopathische systemische Vaskulitis von Blutgefäßen variabler Größe mit episodischem Verlauf [141]
-
Epidemiologie:
-
höchste Prävalenz im mittleren Osten und Asien
-
Geschlechterverhältnis: M : F = 3,5 : 1
-
orbitale Beteiligung extrem selten
-
Ätiologie und Pathogenese:
-
Starke Assoziation zu HLA-B51
-
unspezifische Erhöhung von CRP und BSG [118]
-
Klinik:
-
allgemeine Symptomatik:
-
aphthöse Läsionen der Mundschleimhaut und Genitalien
-
Hautläsionen und intraokuläre Inflammation
-
weitere Organmanifestationen in Gastrointestinaltrakt, Lunge, Muskeln, Gelenken und ZNS [111]
-
bei okulärer Beteiligung:
-
meist Uveitis mit Hypopyon, retinale Vaskulitis
-
selten anteriore ischämische Optikusneuropathie (AION)
-
Cave: okuläre Beteiligung erfordert aufgrund der hohen Morbidität besondere Beachtung
-
bei orbitaler Beteiligung (einzelne Fallberichte):
Befall der extraokulären Muskulatur (EOM) bzw. der Tränendrüse (jedoch ohne bioptische Sicherung) [142]
-
Diagnostik:
-
Bestimmung von HLA-B51, CRP und BSG [118]
-
Diagnose eines orbitalen Befalls anhand der Kombination von klinischen Symptomen und Laborparametern, passend zum Morbus Behçet und der orbitalen Entzündung [118]
-
Therapie:
-
First-Line-Therapie: Kortikosteroide
-
Second-Line-Therapie: Azathioprin, Ciclosprorin, Infliximab oder Adalimumab; in schweren Fällen mit visusbedrohender Retinitis auch Kombinationstherapie möglich [143]
-
aktuelle Studienergebnisse: Einsatz von IL-1-Inhibitoren (Anakinra, Canakinumab) vielversprechend [144]
5.2.3.3.3 Kawasaki-Syndrom
-
Definition:
systemische Autoimmunvaskulitis mit bevorzugtem Befall mittelgroßer Gefäße, insbesondere der Koronargefäße
-
Epidemiologie:
-
Kinder < 5 Jahre
-
Inzidenz: in Japan 240/100.000, in kaukasischer Population 9/100.000 [145]
-
Ätiologie und Pathogenese:
histopathologisch sechs typische Läsionen: Endotheldegeneration, nekrotisierende Arteriitis, granulomatöse Entzündung, Mediadegeneration, Narbenbildung, Ausbildung von Aneurysmen [118]
-
Klinik:
-
Kardinalsymptome: Fieber (> 5 Tage), Palmar- und Plantarerythem, polymorphes Exanthem, bilaterale konjunktivale Injektion und Chemosis, Mundschleimhauterythem, cervikale Lymphadenitis, Aneurysmen der Koronargefäße
-
ophtalmologische Symptome:
-
bilaterale non-purulente Konjuktivitis in 80% der Fälle mit konjunktivaler Injektion und Chemosis
-
anteriore Uveitis [145]
-
orbitale Inflammation (vereinzelte Fallberichte) einmal bioptisch gesichert, ansonsten durch Diagnosestellung im Kontext weiterer Symptome der Erkrankung [146] [147] [148]
-
Diagnostik:
-
typische klinische Konstellation
-
wichtig: kardiologische Diagnostik mit Herz-ECHO, ggf. Koronarangiografie
-
Labor: unspezifische Erhöhung von BSG, CRP, alpha-1-Antitrypsin, Thrombozyten
-
Therapie:
-
Standard: hochdosierte i.v.-Gabe von Immunglobulinen plus Aspirin zur Symptomreduktion und Prävention von Aneurysmen der Koronararterien [118]
-
therapierefraktäre Fälle: Infliximab [149]
-
Einsatz von Kortikosteroiden kontrovers diskutiert (Einsatz nur bei therapierefraktären Fällen) [147]
5.2.3.3.4 Kollagenosen-assoziierte Vaskulitiden bei Systemischem Lupus erythematodes, Rheumatoider Arthritis (RA), Dermatomyositis (DM)
-
Definition:
-
Vaskulitis als typisches Merkmal bei Patienten mit Kollagenosen
-
zumeist Affektion kleinerer Gefäße in Form einer leukozytoklastischen Vaskulitis (perivaskuläre Infiltration mit apoptotisch zerfallenden neutrophilen Granulozyten) [59] [118]
-
Epidemiologie:
-
Vorkommen bei ca. 30–40% der SLE-Patienten und 5–15% der RA-Patienten [59] [118]
-
okuläre Beteiligung bei 20% der Patienten mit SLE
-
orbitale Beteiligung wesentlich seltener [150]
-
Ätiologie und Pathogenese:
immunkomplexvermittelte Pathogenese
-
Klinik:
-
häufig: kutane Läsionen, Befall der Mesenterial-, Koronar- und Pulmonalarterien, der Arterien der unteren Extremitäten und des ZNS
-
okuläre Beteiligung: zumeist Retinopathie, Keratokonjunktivitis sicca, Uveitis
-
vereinzelt Publikationen mit Symptomen einer orbitalen Entzündung (Infarzierung, Myositis, Exophthalmus)
-
orbitale Entzündungssymptome zusammen mit entsprechenden Allgemeinsymptomen und typischen serologischen Befunden bei SLE und RA (erhöhte ANA und ds-DNS-AK) [151] [152] [153]
-
sehr selten orbitale Beteiligung bei Dermatomyositis: Fallbericht einer verzögerten, bilateralen orbitalen Beteiligung mit Exophthalmus durch Vergrößerung der EOM, assoziiert mit Muskelschwäche und typischen EMG-Veränderungen (ohne bioptischen Nachweis) [154]
-
Therapie:
-
SLE und RA: Glukokortikoide i. v. oder p. o.; alternativ Azathioprin und Biologika (Rituximab, Infliximab, Belimumab)
-
DM: Glukokortikoide i. v.; auch Immunglobuline als First-Line-Therapie sowie Methotrexat und Azathioprin beschrieben [118]
5.2.4 Granulomatöse Entzündungen im Bereich der Orbita
5.2.4.1 Sarkoidose
Die Sarkoidose ist eine chronisch entzündliche Systemerkrankung. Der Begriff „sarkoid“ wurde durch Caeser W. Boeck eingeführt, da die Läsionen histologisch Sarkomen ähneln, jedoch benigne sind. Klassisch sind ein Befall der Lungen und eine bihiläre Lymphadenopathie sowie Haut- und Augenlidläsionen [155] [156] [157].
-
Ätiologie und Pathogenese:
-
gesteigerte Entzündungsreaktion auf bislang nicht genauer identifizierte Pathogene mit gesteigerter zellulärer Immunantwort und Ausbildung nicht verkäsender Granulome
-
genetische Komponente (familiäre Häufung)
-
Epidemiologie:
-
Häufung zwischen dem 30. und 40. Lebensjahr
-
Frauen sind häufiger betroffen
-
in ca. 30% der Fälle limitierte orbitale Sarkoidose mit isoliertem Befall orbitaler Strukturen [102] [158]
-
Lokalisation:
-
okuläre Beteiligung (umfasst die häufigsten extrapulmonalen Symptome): Konjunktivitis, Uveitis, chorioretinale Läsionen, Glaskörperveränderungen
-
orbitale Beteiligung als Erstmanifestation möglich: zumeist Tränendrüse (oft bilateral), weiterhin orbitales Fettgewebe, Sehnervenscheide (als Teil einer Neurosarkoidose), ableitende Tränenwege oder Augenmuskeln [155] [156] [159] [160] [161]
-
Klinik:
-
je nach betroffener Struktur palpable Tumormasse, Lidschwellung, Ptosis, Diplopie, Exophthalmus, Epiphora, Sicca-Symptome oder schmerzhafte Ophthalmoplegie [156] [161]
-
bei entsprechender Größe und Lokalisation: Zentralarterienverschluss und Visusverlust möglich [159] [161]
-
Diagnostik:
-
Biopsie zur Diagnosestellung essentiell; durch histologische Ähnlichkeit mit anderen Läsionen wie idiopathischer orbitaler Entzündung oder lymphoproliferativen Erkrankungen Diagnosestellung schwierig
-
Serologie: ACE- und Leberwerte; unauffällige serologische Befunde möglich
-
Bildgebung:
-
MRT: Tränendrüse bei Befall diffus vergrößert mit homogenem Enhancement, weitere Manifestationen als schlecht abgrenzbare Raumforderung, Verdickung der Optikusscheide oder diffus vergrößerte Muskulatur
-
Röntgen-Thorax zur pulmonalen Mitbeurteilung Abklärung
-
Therapie:
-
Kortikosteroide i. v. zur Verbesserung der funktionellen Prognose (von zentraler Bedeutung)
-
interdisziplinäre Betreuung mit Rheumatologen wichtig
-
chirurgische Exzision oder Debulking evtl. erforderlich, allerdings nur in Kombination mit vorheriger Systemtherapie [159] [161]
-
bei therapierefraktären Fällen oder Rezidiven: immunsuppressive Kombinationstherapie, z. B. mit Methotrexat, Azathioprin oder Biologika wie Infliximab oder Adalimumab [156] [161]
5.2.4.2 Melkersson-Rosenthal-Syndrom
-
Definition:
neuromukokutane granulomatöse Dermatose
-
Epidemiologie:
-
Inzidenz 0,08% [162]
-
Erstmanifestation meist im jungen Erwachsenenalter
-
Ätiologie:
autosomal-dominanter Erbgang mit variabler Expression [163]
-
Klinik:
-
klassische Symptomtrias: Lingua plicata, Cheilitis granulomatosa, periphere Fazialisparese; mono- oder oligosymptomatisches Erscheinungsbild wesentlich häufiger
-
häufig diffus-ödematöse Gesichtsschwellungen, selten isoliert im Bereich der Augenlider (hierdurch Diagnosestellung schwierig, besonders beim Fehlen weiterer Charakteristika, Bildgebung und Biopsie können Abgrenzung erleichtern)
-
selten Beteiligung weiterer Hirnnerven [162] [163] [164] [165] (zum Beispiel Diplopie durch Okulomotoriusparese [162] [166])
-
Therapie:
-
intraläsional und systemisch Glukokortikoide
-
Exzision und plastisch-rekonstruktive Gesichtschirurgie bei funktionellen sowie ästhetischen Problemen [59]
-
systemische Therapieansätze mit Immunsuppressiva (z. B. Azathioprin), Hydroxychloroquin und Thalidomid [162] [163] [167]
-
Prognose:
schwankender chronischer Krankheitsverlauf mit spontanen Remissionen und Rezidiven
5.2.4.3 Fremdkörpergranulom und Idiopathisches Lipogranulom
-
Definition:
durch Fremdkörper hervorgerufene granulomatöse Entzündungsreaktion
Einteilung nach der Art des Fremdkörpers:

-
rupturierte Dermoidzyste ([Abb. 13a–d])
-
häufigste intrinsische Ursache für Fremdkörpergranulome (zum Teil mit entzündlicher Begleitreaktion)
-
im CT zumeist inhomogene Läsion mit unregelmäßiger Begrenzung
-
Therapie durch Exzision (möglichst komplette Entfernung ohne Verletzung von Nachbarstrukturen; bei verbliebenen kleinen fibrotischen Anteilen Behandlung mit intraläsionaler Kortikosteroidgabe
-
pflanzliche Fremdkörper
-
weitere Quelle für granulomatöse Entzündungsreaktion, genaue Anamnese wichtig
-
schleichender Verlauf und Fistulation möglich
-
paraffininduziertes sklerotisches Lipogranulom
-
zumeist Folge von Applikation fettender Nasensalbe bei endoskopischen NNH-Eingriffen (mit intraoperativer Verletzung der Lamina papyracea) [168] [169]
-
akzidentelles Austreten von intraokularem Silikonöl im Rahmen ophthalmologischer Eingriffe
-
nach Eigenfettinjektion in der plastischen Chirurgie
-
idiopathisches Lipogranulom
-
bei Fehlen eines auslösenden Ereignisses
-
nur Einzelfälle in der aktuellen Literatur beschrieben, Äthiologie unklar
-
histologisch orbitale Fettgewebsnekrose [170]
-
Klinik:
-
Lidschwellung
-
subkutan palpable Knoten
-
bei raumfordernder Läsion: Exophthalmus, Einschränkungen der Okulomotorik und Bulbusdeviation möglich
-
Diagnostik:
-
klassische Entzündungszeichen durch begleitende Vaskulitis [168] [171] [172]
-
CT oder/und MRT: teilweise unscharf begrenzte, teils diffus konfigurierte Läsion [171]
-
Therapie:
-
möglichst vollständige chirurgische Entfernung mit anschließender systemischer Glukokortikoidgabe am effektivsten [170]
-
optional: Triamcinolon-Injektion oder Debulking
-
Prognose:
sowohl spontane Remission als auch Rezidive möglich [171] [172] [173] [174]
5.2.5 Sjögren-Syndrom (SS)
-
Definition:
-
chronische Autoimmunerkrankung, charakterisiert durch eine Entzündung der Tränen- und Speicheldrüsen sowie der Epithelien
-
Keratokonjunktivitis sicca als eines der führenden diagnostischen Kriterien (Inflammation der akzessorischen konjunktivalen Tränendrüsen) [61]
-
Epidemiologie:
-
Altersgipfel 4.–5. Lebensdekade
-
v. a. Frauen betroffen
-
Einteilung:
-
primäres SS: eigenständiges Krankheitsbild mit okulären und oralen Symptomen (Sicca-Syndrom)
-
sekundäres SS (häufiger): im Rahmen anderer Autoimmunerkrankungen (rheumatoide Arthritis, systemischer Lupus erythematodes, systemische Sklerose)
-
Ätiologie und Pathogenese:
-
Umwelt- und hormonelle Faktoren sowie genetische Prädisposition
-
lymphozytäre Infiltration und Sklerose der Tränen- und Speicheldrüsen durch T-Zellen und Autoantikörper; B-Zellen vermutlich ebenfalls von pathogenetischer Bedeutung [175]
-
Klinik:
-
langsam progrediente, schmerzlose Schwellung der Tränendrüse (und evtl. der Speicheldrüsen)
-
im weiteren Verlauf Drüsenatrophie, häufig bilateral, Xerostomie und Xerophthalmie (eher im atrophen Stadium)
-
weitere okuläre und orbitale Symptome: Uveitis, Episkleritis/Skleritis, Optikusneuropathie[155] [176], Myositiden der EOM [177]
-
extraglanduläre Symptome: Müdigkeit, Schmerzen, Polyneuropathie, vaskulitische Hautläsionen, pulmonale Veränderungen, muskuloskelettale, renale und gastrointestinale Manifestationen [175]
-
Diagnostik:
-
CT/MRT: orbital vergrößerte Tränendrüsen mit Enhancement, Abgrenzung zu lymphoproliferativen Erkrankungen schwierig [178]
-
Labor: Erhöhung spezifischer antinukleärer Antikörper (SS-A und SS-B, letzterer hochspezifisch für das SS)
-
Biopsie: Sicherung der Diagnose und Ausschluss eines Lymphoms (SS-Patienten haben 20-fach erhöhtes Risiko für Lymphome, insbesondere im Kopf-Hals-Bereich [175] [179] [180])
-
Therapie:
-
interdisziplinäres Therapiekonzept notwendig, da SS relativ resistent gegen die medikamentöse Behandlung mit Kortikosteroiden und weiteren Antirheumatika ist
-
symptomatische Therapie der Sicca-Symptomatik durch Ophthalmologen und systemische rheumatologische Basismedikation aus Steroiden und Immunsuppressiva, Einsatz von Biologika, z. B. Rituximab, v. a. bei SS mit extraglandulärer Manifestation empfohlen [181]
5.2.6 Kimura-Syndrom
-
Definition:
-
benigne chronisch-inflammatorische Erkrankung des Subkutangewebes unklarer Genese, auch als angiolymphoide Hyperplasie mit Eosinophilie bezeichnet
-
Erstbeschreibung 1948 durch Kimura [182]
-
Epidemiologie:
-
v. a. bei Männern asiatischer Herkunft zwischen dem 20. und 30. Lebensjahr
-
Befall der Orbita selten
-
Histopathologie:
umschriebene Läsion mit einer Mixtur aus Blutgefäßen mit plumpen Endothelzellen und Zonen polymorpher entzündlicher Infiltrate mit reichlich Eosinophilen, regelrecht aufgebauten multiplen Lymphfollikeln und Ausbildung eosinophiler Abszesse [182] [183] [184] [185]
-
Klinik:
-
inflammatorische Angiome im Kopf-Hals-Bereich mit regionaler Lymphadenitis
-
Befall von Speicheldrüsen sowie Mund- und Nasenschleimhaut möglich
-
Orbita (selten): gut abgrenzbare Raumforderung der Augenlider, Tränendrüse oder anterioren Orbita [184] [186], Exophthalmus, Visuseinschränkung und Doppelbilder
-
Diagnostik:
erhöhtes Serum-IgE und Bluteosinophilie typisch
-
Therapie:
-
Glukokortikoide systemisch und topisch (darunter Tumormassenreduktion und Spontanremissionen möglich)
-
lokale Exzision bei gut abgrenzbarer unifokaler Läsion Therapie der Wahl
-
bei inkompletter Exzision: adjuvante Radiotherapie zur lokalen Kontrolle [184] [186] [187].
5.2.7 Histozytäre Erkrankungen
Orbitale xanthogranulomatöse Erkrankungen sind durch Proliferation von Histiozyten charakterisiert und werden als Non-Langerhans-Zell-Histiozytosen zusammengefasst. Demgegenüber hat die Langerhans-Zell-Histiozytose ihren Ursprung in dendritischen Zellen, das Rosai-Dorfman-Syndrom in Monozyten/Makrophagen, sie werden im Folgenden gesondert aufgeführt.
5.2.7.1 Non-Langerhans-Zell-Histiozytosen
5.2.7.1.1 Juveniles Xanthogranulom

-
Definition:
-
benigne proliferative kutane Erkrankung, hauptsächlich mit Befall von Augen und Haut
-
primärer Befall der Orbita mit Proptosis sehr selten
-
zahlreiche Differenzialdiagnosen im Kindesalter
-
Diagnostik: Bestätigung der Diagnose mittels Biopsie notwendig
-
Therapie und Prognose:
-
meist Spontanremission
-
bei symptomatischen Patienten mit nicht resektabler Läsion Einsatz von Kortikosteroiden und Chemotherapeutika
5.2.7.1.2 Adult-onset Xanthogranulom (AOX) und Adult-onset Asthma mit periokulären Xanthogranulom (AAPOX)
-
Ätiologie und Pathogenese:
-
unklar, vermutlich autoimmune Genese mit Ablagerung von Immunkomplexen und nachfolgender Fremdkörperreaktion im Gewebe
-
Grund der Affinität für orbitale Adnexstrukturen bisher unklar [188]
-
Klinik:
-
gelbliche Infiltrate im Bereich von Konjunktiva und Lidern (Xanthelasmen)
-
bilaterales Auftreten möglich
-
Diagnostik:
-
Bestätigung der Diagnose durch Biopsie: histologisch lipidbeladene Makrophagen (Xanthoma-Zellen) in der Dermis und den tieferen Gewebsschichten, weiterhin Fremdkörperriesenzellen und Touton-Riesenzellen
-
Assoziation mit hämatologischen Erkrankungen (lymphoproliferative Läsionen, Hepatosplenomegalie) möglich, daher gründliche körperliche Evaluation
-
Orbitale Ausdehnungsbestimmung mittels CT: Infiltration des orbitalen Fetts, der EOM und der Tränendrüse
-
bei Assoziation mit Asthma Benutzung des Terminus „Adult-onset Asthma mit periokulärem Xanthogranulom (AAPOX)“ [189]
-
Therapie:
-
chirurgisches Debulking und plastische Rekonstruktion
-
Bestrahlung
-
systemische Kortikosteroide und weitere Immunsuppressiva ([Abb. 14a, b])
-
intraläsionales Triamcinolon sehr effektiv [190]
-
Prognose:
-
progressive Erkrankung
-
ohne Therapie Erblindung
5.2.7.1.3 Nekrobiotisches Xanthogranulom
-
Epidemiologie:
seltene Erkrankung bei Patienten in der 5.–6. Lebensdekade
-
Pathogenese:
-
Fremdkörperriesenzell-Reaktionen durch Ablagerung von Lipid-Serum-Immunglobulin-Komplexen in der Haut[188]
-
Nekrobiosis als histopathologisches Hauptmerkmal: eosinophile Degeneration von Kollagen in einer granulomatösen Entzündungsreaktion mit Schaumzellen, Fremdkörperriesenzellen, Touton-Riesenzellen und Lymphozyten
-
Lokalisation:
-
Entwicklung von Infiltraten an Augenlidern und okulären Adnexstrukturen sowie an den Extremitäten
-
auch Befall innerer Organe und hämatologische Neoplasien möglich [61]
-
Klinik:
rötlich-gelbe Läsionen oder fleischfarbene Knötchen mit Ausdehnung bis in die anteriore Orbita mit nachfolgender Proptosis, Ptosis sowie Narbenlagophthalmus
-
Therapie:
-
viele unterschiedliche Therapiealternativen in Anwendung, inklusive Chirurgie, Radiatio, Plasmapherese, intraläsionale und systemische Steroidgabe sowie Zytostatika
-
Fallberichte zum erfolgreichen Einsatz von Thalidomid in Kombination mit Dexamethason [191]
-
insgesamt Überlegenheit der medikamentösen Therapie gegenüber der Radiatio und dem chirurgischen Debulking
-
Prognose:
-
Befundprogress nach Biopsie oder Debulking möglich
-
lebenslange Nachsorge bei erhöhtem Risiko für hämatologische Malignome erforderlich
5.2.7.1.4 Erdheim-Chester-Krankheit
-
Definition:
-
seltene Form der Non-Langerhans-Zellhistiozytose mit progressivem Verlauf, gekennzeichnet durch Knochenschmerzen, retroperitoneale Fibrose und Infiltrationen im Gesichtsbereich
-
pathologisch charakteristisch: weitverbreitete Infiltration von schaumigen, (lipidbeladenen) Non-Langerhans-Zell-Histiozyten
-
Epidemiologie:
-
Erstmanifestation meist zwischen 5. und 7. Lebensdekade
-
vorrangig bei Männern
-
seit 1930 über 600 Fälle beschrieben [192]
-
Ätiologie:
-
nicht vollständig geklärt
-
Assoziation mit verschiedenen immunologischen Erkrankungen als Hinweis auf abnorme Interaktion zwischen T-Zellen und Makrophagen (unkontrollierte Aktivierung der Makrophagen durch Funktionsstörung von NK-T-Zellen oder zytotoxischen T-Zellen)
-
Klinik:
-
charakteristisch: Knochenschmerzen, v. a. in der distalen unteren Extremität durch eine Osteosklerose der langen Röhrenknochen
-
häufig bilateraler Befall der Orbita: Xanthogranulome mit teilweise tiefer retrobulbärer Infiltration, dadurch neben Exophthalmus auch Ophthalmoplegie und kompressive Optikusneuropathie möglich
-
Allgemeinsymptome wie Fieber und Gewichtsverlust
-
schwere Störungen wie Diabetes insipidus, Kleinhirn- und Pyramidenbahnzeichen, Hirnnervenlähmungen, Nebenniereninsuffizienz, pulmonale Fibrosierung und kardiale Dekompensation möglich [192] [193] [194]
-
variabler Verlauf bis hin zum fulminanten Organversagen
-
Therapie:
-
interdisziplinäre Behandlung bei orbitaler Beteiligung essenziell
-
systemische Therapie mit u. a. Interferon, Glukokortikoiden, Cladribin, Imatinib, Anakinra
-
seit 2012 auch Infliximab und Vemurafenib
-
orbitale Dekompression bei kompressiver Neuropathie des Nervus opticus [192] [194] [195]
-
Prognose:
schlechteste unter allen Non-Langerhans-Zell-Histiozytosen mit Mortalitätsraten > 60% innerhalb von drei Jahren [196]
5.2.7.1.5 Xanthogranuloma disseminatum
-
Definition:
sehr seltene, nicht familiäre, histiozytäre, infiltrative Erkrankung mit primärem Befall von Haut, Schleimhaut sowie Hypothalamus
-
Epidemiologie:
-
etwa 100 berichtete Fälle
-
Erkrankung meist im jungen Erwachsenenalter [197]
-
Gechlechterverhältnis: M : F = 2 : 1
-
Ätiologie und Pathogenese:
-
keine Ursachen oder Risikofaktoren bekannt
-
charakteristisch: Ansammlung von Histiozyten am gesamten Integument und im ZNS mit Akkumulation von Cholesterol und Lipiden, dadurch histologisch schaumiges Aussehen (wie es für xanthomatöse Läsionen typisch ist)
-
Klinik:
-
rötliche kutane Knoten, vor allem an den Oberflächen der Flexoren
-
Befall von Cornea, Konjunktiva und Augenlidern (20%)
-
Dysphagie oder Dyspnoe, bedingt durch Involvierung der oberen Atemwege (40%)
-
Diabetes insipidus, bedingt durch meningealen Befall (40%)
-
Diagnostik:
-
pathognomonisch: Trias aus typischen Hautveränderungen, Dysphagie und Diabetes insipidus [198]
-
Knochenmarksbiopsie und Serumelektrophorese erforderlich zum Ausschluss eines myeloischen Malignoms (bei über 50% der Patienten)
-
histologisch nicht von Erdheim-Chester-Krankheit zu unterscheiden
-
Therapie:
-
kausale Therapie aktuell nicht bekannt
-
Therapieversuche mit zahlreichen verschiedenen Medikamenten, u. a. Kortikosteroiden, Lipidsenkern und vielen Immunsuppressiva; aktuell am vielversprechendsten: Cladribine (Purin-Nukleosid-Analogon) [199]
-
Prognose:
lokale Kontrolle durch chirurgische Exzision oder CO2-Laserablation möglich
5.2.7.2 Langerhans-Zell-Histiozytose
-
Definition:
Spektrum von Erkrankungen, die durch die skelettale oder Weichteilinfiltration mit Langerhans-Zellen charakterisiert sind (Synonym: Langerhans-Zell-Granulomatose, Histiozytosis X) [200] [201]
-
Epidemiologie:
-
Selten, vorwiegend bei Kindern und Jugendlichen
-
Prävalenz: 4–5/1.000.000
-
Befall der Orbita in 25% der Fälle
-
Ätiologie und Pathogenese:
-
unklar, vermutlich klonale Proliferation phänotypisch abnormaler Langerhans-Zellen
-
Expression untypischer Oberflächenproteine und Ablagerung im Gewebe, in dem sonst keine Langerhans-Zellen vorliegen (z. B. Knochen)
-
Formen:
-
unifokale Manifestation: eosinophiles Granulom (häufigste Form)
-
multifokale Manifestation: Hand-Schüller-Christian-Erkrankung mit der Trias aus Exophthalmus, Diabetes insipidus und Osteolysen, sehr selten Letterer-Siwe-Syndrom mit progressivem Befall von Knochenmark und weiteren Organen [202]
-
Klinik:
-
typische orbitale Befunde: Proptosis, Ptosis und Papillenödem, knöcherne Destruktionen der temporalen Orbitaspange
-
zusätzlich entzündliche Begleitreaktion
-
Diagnostik:
-
CT zur Detektion knöcherner Läsionen
-
MRT zur Erfassung der intrakraniellen Ausbreitung [203]
-
Diagnosestellung bei typischer klinischer Läsion, immunhistochemischen Nachweis der Oberflächenmarker CD1a und CD207 sowie elektronenmikroskopischem Nachweis der typischen Birbeck-Granula
-
Differenzialdiagnosen im Kindesalter:
akutes Infektionsgeschehen (Lidödem), entzündlicher Pseudotumor, Dermoidzyste, Rhabdomyosarkom, Ewing-Sarkom, Osteosarkom oder Neuroblastom, Erdheim-Chester-Erkrankung
-
Prognose:
-
besser mit zunehmendem Alter bei Erstdiagnose
-
deutlich schlechter bei multifokalem Befall
-
5-Jahres-Überlebensrate beim Letterer-Siwe-Syndrom trotz moderner Chemotherapeutika max. 50%
-
Therapie:
-
Therapie der Wahl bei isoliertem orbitalen Befall: lokale Exzision
-
intraläsionale Injektion von Kortikosteroiden (rasche Auflösung beschrieben)
-
Low-dose Radiatio bei chirurgisch schwer erreichbaren Läsionen
-
bei inkompletter Entfernung oder Rezidiven: Systemtherapie
-
Prednison bei disseminierter Erkrankung
-
Chemotherapeutika bei schlechtem Ansprechen
-
bei Hypopituitarismus: Hormonsubstitution [61]
5.2.7.3 Sinus-Histiozytose mit massiver Lymphadenopathie (Rosai-Dorfman-Syndrom)
-
Definition:
seltene Erkrankung mit massiver Infiltration von S-100-positiven Histiozyten im Gewebe und den zervikalen Lymphknoten [204]
-
Ätiologie und Pathogenese:
-
möglicherweise virale Genese und genetische Komponente
-
histopathologisch charakteristisch: Emperipolesis mit Phagozytose von Lymphozyten, Erythrozyten und neutrophilen Granulozyten durch große S-100-positive Histiozyten
-
Epidemiologie:
-
Altersgipfel bei Jugendlichen zwischen 15 bis 20 Jahren
-
vermehrtes Auftreten bei Menschen mit schwarzer Hautfarbe [204] [205] [206]
-
Klinik:
-
Erstmanifestation meist durch indolente zervikale Lymphknotenschwellungen mit begleitendem Fieber, Gewichtsverlust und Nachtschweiß
-
extranodalen Manifestationen bei > 40% der Patienten: Auge, Speicheldrüsen, Nase, Knochen, Haut oder ZNS [204] [207], daher systemische Untersuchung notwendig
-
bei massiver Lymphknotenschwellung Kompression der oberen Atemwege mit vital bedrohlichem Zustand möglich [208]
-
bei orbitaler Manifestation: Proptosis, Schwellung von Augenlidern oder Tränendrüse
-
bei intra- oder extrakonalem Befall: kompressive Neuropathie des Nervus opticus und Uveitis möglich [208]
-
Therapie:
-
Erkrankung in der Regel selbstlimitierend; Indikation zur Behandlung v. a. bei drohender Atemwegsverlegung durch plötzliche Größenprogredienz der massiven zervikalen Lymphknotenschwellungen
-
medikamentöse First-Line-Therapie: systemische Hochdosistherapie mit Kortikosteroiden
-
operative Intervention (Exzision oder Debulking) je nach Ausprägungsgrad der orbitalen Manifestation, weitere funktionelle und kosmetische Korrekturen möglich
-
bei idiopathischen Lidödemen systemische Kortikosteroide oder andere Immunsuppressiva
-
bei vital bedrohlicher Verlegung der Atemwege: chirurgische Therapie oder notfallmäßige Radiatio unumgänglich
-
evtl. Chemotherapie bei schwerem orbitalem Befall mit Optikusneuropathie [204] [209]
5.2.8 Morbus Castleman
-
Definition:
seltene lymphoproliferative Erkrankung
-
Einteilung in zwei klinische Formen:
-
unizentrische Form:
-
benigne lokalisierte Hyperplasie lymphatischen Gewebes
-
kurative Behandlung durch Exzision der betroffenen Lymphknoten
-
multizentrische Form:
-
Systemerkrankung mit potenziell lebensbedrohlicher Symptomatik
-
Einteilung in HHV-8-assoziierte Form und idiopathische multizentrische Form (pathogenetisch Zytokindysregulation) [210] [211]
-
Einteilung in drei histologisch Typen:
-
hyalin-vaskulärer Typ (80–90%)
-
Plasmazelltyp (10–20%)
-
seltener Mischtyp
-
Lokalisation:
-
extranodaler Befall der Orbita sehr selten, nur wenige Fälle beschrieben
-
unizentrische Variante mit hyalin-vaskulärer Form in über 90% der orbitalen Fälle
-
Klinik:
-
zumeist indolente Schwellung (Proptosis)
-
in einigen Fällen B-Symptomatik
-
Diagnostik:
-
bildgebende Diagnostik zur orbitalen Befundbeschreibung und zum Ausschluss eines multizentrischen Auftretens empfehlenswert
-
Therapie:
-
bei multizentrischer Variante: interdisziplinäre Behandlung anstreben
-
monoklonale Antikörper, ggf. in Kombination mit Zytostatika [211] [213] [214]
5.3 Konklusion
Die große Bandbreite der möglichen Diagnosen und die häufig überlappende klinische und radiologische Präsentation orbitaler Entzündungszeichen unterstreichen die entscheidende Bedeutung von Biopsien, um die Ursachen der Läsionen zu identifizieren. Neben den mannigfaltigen infektiösen Entzündungen, welche seit jeher aufgrund der Akuität des Krankheitsbildes und den potenziellen schwerwiegenden Komplikationen große Bedeutung haben, präsentieren sich auch autoimmune Erkrankungen oft initial in der Orbita und müssen serologisch und systemisch sehr genau abgeklärt werden. Von den nicht infektiösen inflammatorischen Läsionen ist die idiopathische orbitale Entzündung am häufigsten. IgG4-assoziierte Erkankungen haben in den letzten Jahren als Ursache orbitaler Entzündungen an Bedeutung gewonnen. Wegen ihrer Ähnlichkeit zu lymphoproliferativen Erkrankungen sollte man bei der Beurteilung nicht infektiöser Entzündungen der Orbita besonders achtsam sein. Histiozytäre Erkrankungen bleiben wichtige, wenn auch seltene Erkrankungen, welche Entzündungen und Fibrosierungen der Orbita hervorrufen, aber auch zu schwerem Organversagen führen können.
6. Degenerative Erkrankungen
6.1 Orbitale Amyloidose
Die Amyloidose beschreibt eine heterogene Gruppe von Erkrankungen, bei denen es zur extrazellulären Akkumulation von unlöslichen Proteinen in β-Faltblattstruktur kommt. Dabei kann die Organfunktion durch Kompression oder direkte Zytotoxizität deutlich gestört werden. Eine Amyloidose kann primär vorliegen oder sekundär als Folge einer anderen Erkrankung. Eine genetische Disposition ist beschrieben [215]. Es handelt sich um eine seltene, systemisch oder lokalisiert auftretende Erkrankung.
-
Lokalisation:
-
in allen Strukturen der Orbita und des okulären Gewebes Ablagerung des Amyloids möglich [216] [217] [218]
-
Fälle mit Beteiligung von Tränendrüse, Lidern, Konjunktiva und Augenmuskeln, einschließlich des Musculus levator palpebrae, beschrieben (siehe [Abb. 15a + b])


-
Klinik:
Ptosis, Exophthalmus, Motilitätseinschränkungen mit Doppelbildern, Bulbusdeviation, Ek- oder Entropium, palpabler Tumor [218]
-
Differenzialdiagnosen:
-
chronisch entzündliche Darmerkrankungen, Tuberkulose, Multiples Myelom
-
Untersuchungen von Herz, Nieren, Gastrointestinaltrakt und ZNS notwendig [215]
-
Diagnostik:
-
Diagnosestellung durch Fehlen typischer Symptome erschwert
-
-
MRT: Ablagerungen inhomogen und KM-anreichernd
-
CT: Kalzifikationen
-
Histopathologie: Amyloid-typische apfelgrün leuchtende Anfärbbarkeit sowie polarisationsmikroskopisch doppelt lichtbrechende Eigenschaften
-
bei lokalisiertem Befall muss die systemische Variante sicher ausgeschlossen werden
-
Therapie:
-
in Abhängigkeit von der jeweiligen Form von systemischer Chemotherapie mit Kortisongabe bis hin zur Transplantation der betroffenen Organe
-
bei lokalisierter orbitaler Amyloidose: operative Entfernung als Therapie der Wahl, evtl. korrigierende Eingriffe z.B. bei Lidfehlstellungen [215] [216] [218] [219].
-
Strahlentherapie zur Rezidivprophylaxe von einigen Autoren empfohlen [216] [220]
-
Prognose:
sehr variabler klinischer Verlauf bei orbitaler Beteiligung, von komplikationslosen lokalisierten Befunden bis zu schwerer Organschädigung mit Visusverlust [215]
6.2 Speicherkrankheiten [221] [222] [223] [224] [225]
Auch Patienten mit Speicherkrankheiten wie dem Morbus Pompe oder Morbus Gaucher haben, insbesondere beim klassischen infantilen Auftreten, eine hohe Prävalenz klinisch signifikanter ophthalmologischer Symptome wie Ptosis, Bulbusmotilitätsstörungen, Strabismus, Myopie und Astigmatismus. Diese ergeben sich unter anderem aus der Akkumulation von Glykogen (Morbus Pompe) bzw. Glukozerebrosid (Morbus Gaucher) insbesondere in der extraokulären Muskulatur und können weitere okuläre Adnexstrukturen befallen.
6.3 Mitochondriale Erkrankungen
6.3.1 Chronisch progressive externe Ophthalmoplegie (CPEO)
-
Definition:
sehr seltene systemische Muskelerkrankung mit langsam progredienter Lähmung aller äußeren Augenmuskeln
-
Epidemiologie:
-
neben der Leberschen hereditären Optikusatrophie die häufigste mitochondriale Erkrankung des Erwachsenenalters mit Augenbeteiligung
-
Prävalenz: etwa 12/100.000 [226]
-
Beginn meist zwischen 20. und 50. Lebensjahr
-
Klinik:
Ptosis häufig erstes Symptom [227]
-
Differenzialdiagnosen:
-
andere mitochondriale Erkrankungen, beispielsweise das Kearns-Sayre-Syndrom (ebenfalls Ptosis und externe Ophthalmoplegie, Beginn meist vor dem 20. Lebensjahr, zusätzlich Reizleitungsstörungen am Herzen, zerebelläre Ataxie und/oder Liquoreiweißerhöhung)
-
MELAS-Syndrom, (okuläre) Myasthenia gravis, okulopharyngeale Muskeldystrophie
-
Diagnose:
-
MRT: Atrophie der extraokulären Muskulatur [228]
-
Skelettmuskelbiopsie: Nachweis zahlreicher „ragged red fibers“ und COX-negativer Fasern
6.3.2 Leigh-Syndrom
-
Definition: neurodegenerative Erkrankung (Mitochondriopathie) bei Kindern
-
Epidemiologie:
-
häufigste pädiatrische Präsentation einer Mitochondriopathie
-
Auftreten zumeist im 2. Lebensjahr
-
Prävalenz: ca. 1/40.000 [229]
-
in seltenen Fällen auch Auftreten im Erwachsenenalter möglich [230]
-
Ätiologie:
-
genetisch sehr heterogen, bislang Identifikation von Mutationen in 75 Genen [231]
-
Klinik:
-
sehr variable Symptomatik
-
im Rahmen eines Infekts zunächst Entwicklung von neurologischen Symptomen wie Ataxie und Dystonie, aber auch ophthalmologischen Symptomen, v. a. Strabismus, Pigmentretinopathie, Optikusatrophie, Ptosis und Nystagmus [232]
-
später Symptome an weiteren Organen wie Herz, Leber, Nieren oder Gastrointestinaltrakt
-
Prognose:
schubartiger Progress, meist mit Tod im 3. Lebensjahr
7. Tumoren der Orbita [2] [201] [233]
7.1 Allgemeiner Teil
Orbitale Neoplasien sind mit einer Inzidenz von 6 bis 10 pro 1 Million Einwohner selten, umfassen aber eine Vielzahl verschiedener Entitäten, was bereits im Rahmen der Diagnostik zu Schwierigkeiten führen kann. Etwa 60% sind gutartig, 40% bösartig (Erwachsene). Abzugrenzen sind „echte“ orbitale Neoplasien von entzündlichen orbitalen Raumforderungen ohne erkennbare Ursache (idiopathische orbitale Entzündung, früher sogenannte Pseudotumoren). Diese machen etwa 5–7% aller orbitalen Raumforderungen aus und sprechen gut auf Steroide und Immunsuppressiva an. Im Rahmen der Anamnese und Diagnostik sollten die genaue Lokalisation, die Invasivität sowie die Dignität abgeklärt werden. Diagnostik und Therapie erfolgen häufig interdisziplinär.
7.1.1 Symptome
Aufgrund der anatomischen Gegebenheiten kommt es, in absteigender Häufigkeit, typischerweise zu folgenden Symptomen [201]:
-
Exophthalmus / Protrusio bulbi (Hauptsymptom)
-
Bulbusdislokation
-
Motilitätsstörungen, Doppelbilder
-
Schmerzen
-
Visusminderung
-
Gesichtsfeldeinschränkung
Intrakonale Raumforderungen führen eher zu einem axialen Exophthalmus, extrakonale Raumforderungen eher zu einer Bulbusdislokation zur dem Tumor entgegengesetzten Seite (z. B. führt ein Tumor der Tränendrüse zur Dislokation nach nasal unten).
7.1.2 Einteilung und Überblick [2] [201]
Neoplasien der Orbita lassen sich wie folgt unterteilen:
-
Neoplasien epithelialen Ursprungs
-
Neoplasien nicht epithelialen Ursprungs
-
Neoplasien des lymphatischen Gewebes
In jeder Kategorie findet man gutartige und bösartige Vertreter. Einzige epitheliale Struktur in der Orbita ist die Tränendrüse, somit sind primäre epitheliale Neoplasien nur dort zu finden. Vaskuläre Raumforderungen werden teilweise nicht als Neoplasie, sondern als Malformation erachtet und nehmen eine Sonderstellung ein. Sie können sich klinisch dennoch wie „echte“ Neoplasien verhalten.
7.1.3 Allgemeine Therapieprinzipien bei gutartigen Neoplasien der Orbita [2] [201]
Folgende Therapieoptionen bestehen im Allgemeinen:
-
„Watch and wait“ bei asymptomatischen benignen Neoplasien, bei denen keine diagnostische Unsicherheit besteht (z. B. beim zufällig entdeckten Hämangiom)
-
chirurgische Resektion als häufigste Therapieform bei symptomatischer benigner Neoplasie
-
andere Therapiestrategien wie Strahlentherapie oder Chemotherapie bei speziellen Entitäten
Bei symptomatischen, gutartigen Prozessen ist eine gründliche Abwägung zwischen dem zu erwartenden therapeutischen Benefit und dem Risiko eines funktionellen Schadens erforderlich. So ist beispielsweise die Resektion eines Optikusscheidenmeningeoms mit einem hohen Risiko für eine Optikusatrophie assoziiert.
7.2 Spezieller Teil
7.2.1 Neoplasien der Tränendrüse
-
Inzidenz: 0,6–1/1.000.000 Einwohner pro Jahr; etwa 80 Neuerkrankungen pro Jahr in Deutschland
-
Tumoren der Tränendrüse machen etwa 25 % der orbitalen Raumforderungen aus
-
Die Häufigkeitsverteilung von Tumoren der verschiedenen Gewebetypen in der Tränendrüse stellt sich wie folgt dar [237]:
-
epithelialer Ursprung: > 50–65 %
-
lymphatischer Ursprung (Sonderform mesenchymaler Neoplasien): circa 30–35 %
-
mesenchymaler Ursprung oder Metastasen: 10–15%
-
Verhältnis gutartiger zu bösartigen epithelialen Neoplasien annähernd 1 : 1
-
Differenzialdiagnosen [233]:
-
entzündliche Erkrankungen
-
akute Dakryozystitis
-
chronische Dakryozystitis
-
idiopathische orbitale Entzündung (IOE, früher „Pseudotumor orbitae“, dakryoadenitische Form, die ca. 20–40% aller IOE ausmacht)
-
strukturelle Veränderungen (z. B. Tränendrüsenzyste)
-
sekundäre Tumoren (nicht vom Drüsengewebe ausgehend, z. B. Metastasen, Schwannome)
-
Bei Verdacht auf einen epithelialen Tumor der Tränendrüse sollte keine Inzisionsbiopsie erfolgen, da dies die Prognose sowohl bei gutartigen wie auch bösartigen Tumoren signifikant verschlechtert (pleomorphes Adenom: 5-Jahres-Rezidivrate von 32% nach Biopsie vs. 3% ohne Biopsie vor der Tumorresektion, zudem können die lokal rezidivierten pleomorphen Adenome entarten; Tränendrüsenmalignom: 5-Jahres-Überlebensrate von 29% nach Biopsie vs. 70% bei initial vollständiger Resektion).
-
Eine möglichst genaue präoperative diagnostische Einordnung, unter anderem anhand der Symptomdauer, des Vorhandenseins bzw. Fehlens von Schmerz und anhand radiologischer Merkmale, ist daher sehr wünschenswert
-
Effektivste und sicherste Therapie bei epithelialen Tumoren ist die vollständige Resektion während der ersten chirurgischen Maßnahme.
-
Bekapselte und gut umschriebene Prozesse sollten ohne vorherige Inzisionsbiopsie vollständig reseziert werden.
-
Der operative Zugang erfolgt über eine laterale Orbitotomie, selten in Kombination mit einem Bügelschnitt.
7.2.1.1 Gutartige Neoplasien der Tränendrüse
[Tab. 3] fasst die wichtigsten gutartigen Neoplasien der Tränendrüse zusammen. Lymphoide Neoplasien werden, auch im Kapitel über bösartige Erkrankungen, aufgrund ihrer speziellen Eigenschaften gesondert aufgeführt, obwohl sie streng genommen zu den Neoplasien mesenchymalen Ursprungs gehören.
|
Art des Gewebes |
Neoplasie |
|---|---|
|
epithelial |
pleomorphes Adenom |
|
Warthin-Tumor |
|
|
Onkozytom |
|
|
Myoepitheliom |
|
|
Zystadenom |
|
|
sklerosierende polyzystische Adenose |
|
|
mesenchymal |
solitärer fibröser Tumor |
|
Myxom |
|
|
fibröses Histiozytom |
|
|
lymphoid |
reaktive lymphoide Hyperplasie |
7.2.1.1.1 Gutartige epitheliale Neoplasien der Tränendrüse
7.2.1.1.1.1 Pleomorphes Adenom [233] [234] [236] [238] [239] [240] [241] [242] [243] [244]
-
Epidemiologie:
-
häufigster epithelialer Tumor der Tränendrüse (> 50 %)
-
ca. 20% aller Tränendrüsentumoren [237]
-
Manifestation in 4.–5. Lebensdekade
-
m : w = 1 : 1
-
-
typische Klinik:
-
sehr langsames Wachstum
-
Bulbusverlagerung nach nasal unten (95%), Motilitätseinschränkungen ggf. mit Doppelbildern (40%), Epiphora
-
Diagnostik:
-
CT als Diagnostikum der Wahl
-
durch MRT nur wenig Zusatzinformation (hyperintenses Signal in der T2-Wichtung)
-
extrakonale, rundlich-ovale, scharf abgrenzbare Raumforderung im lateralen oberen Quadranten der Orbita, inhomogene Binnenstruktur, verdrängendes Wachstum, Aufweitung der knöchernen Fossa glandulae lacrimalis (81% im CT), Verkalkungen können Zeichen maligner Transformation sein
-
Sonografie: gut abgrenzbare, echoarme Raumforderung mit dorsaler Schallverstärkung, wenig Binnenechos; in der Farbduplexsonographie nur im Bereich der Kapsel Vaskularisation und Perfusion nachweisbar
-
-
weiteres Diagnosekriterium: Score zur Unterscheidung zwischen pleomorphem Adenom und Karzinom der Tränendrüse nach Rose und Wright [242] ([Tab. 4])
-
Biopsie:
-
Gefährlichkeit der Inzisionsbiopsie von einigen Autoren angezweifelt, von den meisten jedoch bestätigt
-
Nutzen und Risiken einer Feinnadelpunktionszytologie umstritten
-
-
Therapie:
-
komplette Tumorexstirpation ohne Eröffnung der Tumorkapsel mit einem Randsaum aus Drüsengewebe (Zugang: laterale Orbitotomie, palpebraler Drüsenanteil sollte erhalten bleiben!)
-
nach vorher stattgehabter Biopsie: En-bloc-Resektion der Hautnarbe mit darunter liegendem Biopsiekanal sowie der Tränendrüse
-
-
Prognose:
|
Eigenschaft |
Punktwert −1 |
Punktwert +1 |
|---|---|---|
|
Dauer der Symptome |
< 10 Monate |
> 10 Monate |
|
persistierender Schmerz |
+ |
– |
|
Sensibilitätsstörung |
+ |
– |
|
gut abgrenzbar, rund-oval (CT) |
– |
+ |
|
Wachstum entlang orbitaler Strukturen (CT) |
+ |
– |
|
Verkalkungen (CT) |
+ |
– |
|
knöcherne Destruktionen (CT) |
+ |
– |
|
Verhältnis von Symptomdauer zu Tumorgröße |
großer Tumor mit kurzer Symptomdauer |
kleiner Tumor mit langer Symptomdauer |
−8 bis +2 Punkte → eher karzinomverdächtig → Inzisionsbiopsie +3 bis +8 Punkte → Verdacht auf pleomorphes Adenom → Tumorentfernung in toto (Inzisionsbiopsie kontraindiziert)
bei unvollständiger Resektion und Inzisionsbiopsien oder bei Nichtbehandlung Risiko von Rezidiven (32% innerhalb von 5 Jahren nach Inzisionsbiopsie vs. 3% nach vollständiger kapselerhaltender Resektion) bzw. maligner Entartung (20% innerhalb von 30 Jahren)
7.2.1.1.1.2 Warthin-Tumor (Cystadenolymphom) [245] [246]
-
Gehört zu den monomorphen Adenomen
-
Synonyme: Adenolymphom, Zystadenolymphom, papilläres Cystadenoma lymphomatosum (um eine Verwechslung mit malignen Lymphomen oder dem Lymphadenom zu vermeiden, sollte jedoch die Bezeichnung Warthin-Tumor verwendet werden)
-
Epidemiologie:
-
zweithäufigster gutartiger Tumor der Glandula parotidea, gelegentlich auch in den periparotidealen Lymphknoten
-
selten in anderen Speicheldrüsen
-
extrem selten in der Tränendrüse (bisher nur < 10 Fälle weltweit beschrieben)
-
Therapie:
vollständige chirurgische Resektion über laterale Orbitotomie
7.2.1.1.1.3 Onkozytom [247] [248] [249] [250] [251] [252] [253] [254]
-
Synonyme: oxyphiles Adenom, onkozytäres Adenom, Hürthle-Zelladenom
-
Epidemiologie:
-
extrem selten in der Tränendrüse (MEDLINE-Analyse der Weltliteratur von 1959 bis 2004 ergab nur fünf gut dokumentierte Fälle [248])
-
häufiger in Speicheldrüsen, Niere, Adenohypophyse, (Neben-)Schilddrüse, Karunkel und Konjunktiva des Auges
-
v. a. ältere Frauen betroffen
-
-
Ätiologie:
Entstehung vermutlich durch eine Mutation der mitochondrialen DNA
-
Histologie:
-
gutartiger, langsam wachsender, von den Epithelien der Drüsenausführungsgänge ausgehender Tumor; Entartung höchst selten
-
aufgebaut aus feingranulierten, großen, eosinophilen, mitochondrienreichen Tumorzellen, den sogenannten Onkozyten („Hürthle-Zellen“)
-
Therapie der Wahl:
vollständige chirurgische Resektion
7.2.1.1.1.4 Myoepitheliom [241] [255] [256] [257] [258] [259]
-
gehört zu den monomorphen Adenomen
-
Epidemiologie:
-
Bildgebung:
-
im CT gut abgrenzbare, bekapselte Raumforderung
-
-
in anderen Lokalisationen klinisch-radiologisch schwer von anderen Weichgewebstumoren oder -sarkomen zu unterscheiden
-
Therapie:
vollständige und intakte Resektion über laterale Orbitotomie, ggf. kombiniert mit Bügelschnitt [255]
-
Nachsorge:
radiologisches Follow-up empfohlen
-
Prognose:
Lokalrezidivrate: 20%
7.2.1.1.1.5 Zystadenom [260] [261] [262] [263]
-
Epidemiologie:
-
Rarität in der Tränendrüse
-
vor allem in Gallengängen Pankreas, Ovarien, Nebenhoden und Nieren
-
selten in den Speicheldrüsen (< 1% der Speicheldrüsentumoren)
-
-
Pathologie:
-
papilläre und muzinöse Subtypen (letztere können entarten)
-
Bajaj et al. (2002): erstmaliger Bericht über eine bilaterales Zystadenom der Tränendrüsen [260]
-
-
Differenzialdiagnose:
papilläres Zystadenom ähnelt histologisch dem papillären Cystadenoma lymphomatosum (= Warthin-Tumor)
7.2.1.1.1.6 Sklerosierende polyzystische Adenose (SPA) [264] [265]
-
Definition:
-
neue Entität in der aktuellen WHO-Klassifikation der epithelialen Speicheldrüsentumoren
-
früher als entzündliche/reaktive Veränderung erachtet, aktuell Bezeichnung als Neoplasie zwar umstritten, jedoch von den meisten Autoren favorisiert
-
-
Synonym: sklerosierendes polyzystisches Adenom
-
Epidemiologie:
-
erster Fallbericht einer SPA der Tränendrüse im Jahr 2013 [264]
-
bisher nur etwa 60 Fälle in der Literatur beschrieben (Befall meist der Glandula parotidea, gelegentlich der kleineren Spreicheldrüsen)
-
-
Differenzialdiagnosen:
pleomorphes Adenom, adenoidzystisches Karzinom, Mucoepidermoidkarzinom, Azinuszellkarzinom (häufig histopathologische Missinterpretation und Verwechslung)
-
Prognose:
-
Rezidiv in 30% der Fälle
-
keine Metastasierung oder krankheitsbedingt erhöhte Sterblichkeit durch SPA bekannt
-
7.2.1.1.2 Gutartige nicht epitheliale Neoplasien der Tränendrüse
7.2.1.1.2.1 Tumoren mesenchymalen Ursprungs
Solitärer fibröser Tumor [266] [267] [268] [269] [270] [271]
-
Epidemiologie:
-
insgesamt sehr seltener Tumor unabhängig von der Lokalisation
-
2,8 Fälle auf 100.000 Patienten im Krankengut der Mayo Clinic [268]
-
im Bereich der Tränendrüse ca. 10 Fallberichte in der Literatur
-
typische Lokalisationen: am häufigsten an der Pleura, seltener Bauch, Becken
-
-
Pathologie:
-
im Bereich der Tränendrüse möglicherweise von periduktalem Bindegewebe ausgehend
-
meist gutartig, bösartige Formen (10−37%) sowie Rezidive und Metastasen kommen jedoch vor
-
langsames, verdrängendes Wachstum
-
-
Differenzialdiagnose:
im Bereich der Tränendrüse ähnliches klinisches und radiologisches Erscheinungsbild wie pleomorphes Adenom
-
Therapie:
-
vollständige Resektion über laterale Orbitotomie
-
keine schlüssige Evidenz für Benefit von Radiotherapie oder Chemotherapie bei Residualtumoren
-
-
Prognose:
-
Nachweis von mikroskopisch nicht tumorfreien chirurgischen Resektionsrändern korreliert mit einer erhöhten Entwicklung von Lokalrezidiven und Fernmetastasen
-
Lokalrezidive und Fernmetastasen werden insbesondere bei Tumoren > 10 cm beschrieben, wie sie im Thorax oder Abdomen vorkommen
-
extrathorakale Lokalisation zeigt erhöhtes Risiko für Lokalrezidive, aber nicht für die Entwicklung von Fernmetastasen
-
7.2.1.1.2.1.1 Myxom [272] [273] [274] [275] [276]
-
Definition:
Neoplasie mesenchymalen/bindegewebigen Ursprungs
-
Epidemiologie:
-
1 Fallbericht in der Tränendrüse
-
sonstige Lokalisationen am Auge: Konjunktiva, Kornea, Orbita
-
andere Lokalisationen: vor allem Herz, daneben Knochen, Haut, Skelettmuskel, Urogenitaltrakt
-
-
Pathologie:
histologisch zellarm und gefäßarm mit reichlich myxoider (schleimähnlicher) hyaluronsäurereicher Matrix
-
Diagnostik:
im CT gut umschriebene, ovaläre isodense Raumforderung
-
Therapie:
-
vollständige chirurgische Resektion (dann exzellente Prognose)
-
Strahlentherapie ohne relevanten Effekt
-
7.2.1.1.2.1.2 Fibröses Histiozytom [261] [277]
-
Epidemiologie:
-
Bericht über ein fibröses Histiozytom der Tränendrüse bei einem elfjährigen Mädchen ohne weitere Fallberichte dazu in der Literatur [277]
-
Häufiger in der Orbita bei Erwachsenen
-
-
Pathologie:
Unterscheidung zwischen gutartigen, lokal aggressiven und bösartigen Formen
7.2.1.1.2.2 Gutartige Tumoren lymphoiden Ursprungs – Reaktive lymphoide Hyperplasie [278]
-
Definition:
-
gutartige lymphoproliferative Erkrankung
-
keine „echte“ Neoplasie, da nicht monoklonal und nicht autonom
-
polyklonale, lymphozytäre Proliferation
-
früher als Pseudolymphom bezeichnet
-
„Atypische lymphoide Hyperplasie“ als Sonderform (Borderline-Läsion zwischen reaktiver lymphoider Hyperplasie und Lymphom, heutzutage meist den Low-grade-B-Zell-Lymphomen zugerechnet)
-
-
Epidemiologie:
-
ca. 6% aller Läsionen der Tränendrüse [279]
-
kommt in ähnlicher Häufigkeit in Konjunktiven, Tränendrüse und sonstigen Lokalisationen der Orbita vor (dort meist extrakonal), selten im Augenlid
-
selten bilateral oder zusätzlich außerhalb der Augenregion (z. B. Parotis, Lunge)
-
-
Klinik:
-
langsam wachsende Raumforderung mit geringer funktioneller Beeinträchtigung, nur selten Rötung oder Schmerzen
-
in der Tränendrüse eventuell palpabel als eher feste, gummiartige Raumforderung mit glatter oder knotiger Oberfläche
-
-
Therapie [237]:
-
Hochdosis-Glukokortikoide und/oder
-
Strahlentherapie (circa 25 Gy)
-
Rituximab bei therapieresistenten Fällen
-
-
Prognose:
maligne Entartung möglich
7.2.1.2 Bösartige Neoplasien der Tränendrüse
[Tab. 5] fasst die bösartigen Neoplasien der Tränendrüse zusammen.
|
Art des Gewebes |
Neoplasie |
|---|---|
|
epithelial |
adenoidzystisches Karzinom |
|
Karzinom im pleomorphen Adenom |
|
|
Adenokarzinom (NOS) |
|
|
Mukoepidermoidkarzinom |
|
|
duktales Karzinom |
|
|
Azinuszellkarzinom |
|
|
Talgdrüsenkarzinom |
|
|
myoepitheliales Karzinom |
|
|
Plattenepithelkarzinom |
|
|
onkozytisches Karzinom |
|
|
polymorphes Adenokarzinom |
|
|
sekretorisches Adenokarzinom |
|
|
nicht epithelial |
Synovialsarkom |
|
granulozytisches Sarkom |
|
|
maligner peripherer Nervenscheidentumor |
|
|
maligner rhabdoider Tumor |
|
|
lymphoid |
extranodales Marginalzonen-B-Zell-Lymphom |
|
follikuläres Lymphom |
|
|
diffus-großzelliges B-Zell-Lymphom |
|
|
Mantelzelllymphom |
7.2.1.2.1.Bösartige Tumoren der Tränendrüse epithelialen Ursprungs
Vergleicht man alle Formen von Karzinomen der Tränendrüsen und der großen Speicheldrüsen, so scheinen Tumore mit ähnlicher Histologie eine ungünstigere Prognose zu haben, wenn sie in der Tränendrüse auftreten. Für die Karzinome der Tränendrüse existiert eine TNM-Klassifikation in der aktuellen Auflage der „TNM-Klassifikation maligner Tumoren“ der UICC. Als regionäre Lymphknoten gelten demnach die präaurikulären, submandibulären und zervikalen Lymphknoten. Die Zuordnung zu den T-Kategorien T1–3 basiert auf der Tumorgröße, wobei je nach Beteiligung von Periost und/oder Knochen weiter in die Unterkategorien a–c unterteilt wird. Eine Beteiligung von Nachbarstrukturen, z. B. Nasennebenhöhlen, Sinus cavernosus oder Gehirn, führt zur Einteilung in die Kategorie T4 [280].
Eine eigene histopathologische Klassifikation für Tränendrüsentumoren existiert nicht. Stattdessen wird die Tränendrüse von vielen Autoren wie eine kleine Speicheldrüse betrachtet und die Tumoren der Tränendrüse werden entsprechend denen der Speicheldrüsen klassifiziert.
[Tab. 6] zeigt einen Vorschlag zur Klassifikation der bösartigen epithelialen Tränendrüsentumoren modifiziert nach Weis et al. in Anlehnung an die WHO-Klassifikation der Speicheldrüsentumoren und mit Aktualisierungen entsprechend deren aktueller Version [237] [261] [265] [285] [286]. Erwähnenswert ist, dass bis vor einigen Jahren primäre Adenokarzinome der Tränen-, aber auch der Speicheldrüsen nicht weiter unterteilt wurden. Inzwischen weiß man, dass es sich um eine Gruppe von Tumoren mit unterschiedlicher Morphologie, unterschiedlichem biologischem Verhalten und damit auch unterschiedlicher Prognose handelt.
|
Klassifikation |
Neoplasie |
|---|---|
|
low-grade |
Karzinom im pleomorphen Adenom (bei minimal-invasivem Karzinom [≤ 1,5 mm]) |
|
polymorphes Adenokarzinom |
|
|
Mukoepidermoidkarzinom Grad 1/2 |
|
|
epithelial-myoepitheliales Karzinom |
|
|
Azinuszellkarzinom |
|
|
Basalzell(adeno)karzinom |
|
|
muzinöses Adenokarzinom |
|
|
Klarzellkarzinom |
|
|
Zystadenokarzinom[ 281] |
|
|
sekretorisches Karzinom |
|
|
high-grade |
Karzinom (Adenokarzinom oder adenoidzystisches Karzinom) im pleomorphen Adenom (bei minimal-invasivem Karzinom [> 1,5 mm]), „malignant mixed tumor“ |
|
adenoidzystisches Karzinom (not otherwise specified) |
|
|
Adenokarzinom (not otherwise specified) |
|
|
Mukoepidermoidkarzinom Grad 3 |
|
|
duktales Adenokarzinom |
|
|
Plattenepithelkarzinom |
|
|
Talgdrüsenkarzinom |
|
|
myoepitheliales Karzinom |
|
|
onkozytisches Karzinom |
|
|
lymphoepitheliales Karzinom |
|
|
Karzinosarkom (im pleomorphen Adenom, “true malignant mixed tumor”) [282] |
|
|
neuroendokrines Karzinom[ 283] |
|
|
Merkelzell-Karzinom[ 284] |
|
|
andere seltene und nicht klassifizierbare Karzinome |
|
|
Entdifferenzierung in einem der oben genannten Karzinome |
7.2.1.2.1.1 Adenoidzystisches Karzinom (ACC) [279] [287] [288] [289] [290] [291] [292] [293] [294] [295]
-
Epidemiologie:
-
Pathologie:
-
perineurale Invasion des Tumors als typisches Zeichen
-
häufig hämatogene Metastasierung (typischer als die regionäre Lymphknotenmetastasierung!) – auch noch nach vielen Jahren (v. a. in Lunge, daneben Knochen, Leber und Gehirn)
-
-
Klinik:
-
Bulbusdislokation, Proptosis, S-förmige Ptosis, Doppelbilder, Epiphora
-
Schmerzen (in 38–79% der Fälle) und Hypästhesie im frontotemporalen Bereich (Hinweis auf Vorliegen eines aggressiven Tumors)
-
Symptomdauer bei Erstvorstellung typischerweise < 6 Monate
-
-
Diagnostik:
-
hochauflösende CT: Knochenarrosion, irreguläre Berandung der Raumforderung, eventuell fokale Verkalkungen innerhalb des Tumors
-
MRT: beste Möglichkeit zur Beurteilung einer Beteiligung von Sinus cavernosus, Hirn und Knochenmark
-
-
Therapie:
-
noch immer Unklarheit und Uneinigkeit hinsichtlich der optimalen Therapie; meist Resektion mit Sicherheitsabstand und adjuvante Strahlentherapie
-
Operative Möglichkeiten:
-
je nach Befundausdehnung lokale Resektion, Exenteratio oder radikale Exenteratio (mit Resektion von Orbitadach, lateraler Orbitawand und Anteilen des Musculus temporalis)
-
radikale Chirurgie ohne erkennbaren Vorteil für die lokale Kontrolle bei Tumoren bis T2, Exenteratio bei Tumoren ab T3 wohl von Vorteil [297]
-
(elektive) Neck Dissection: Die Rate der regionären Lymphknotenmetastasen (einschließlich der intraparotidealen Lymphknoten!) gilt bei ACC (insbesondere der Tränendrüse) als gering [297] [298]. Die elektive Neck Dissection erscheint daher in den meisten Fällen nicht gerechtfertigt (sollte aber in fortgeschrittenen Stadien, beim soliden histologischen Subtyp oder bei hochgradiger Entartung diskutiert werden) [298]. Bei ACC der Speicheldrüsen liegt die lymphogene Metastasierungsrate wohl höher, die elektive Neck Dissection führt dort bei fortgeschrittenem Stadium zu einer höheren Überlebenswahrscheinlichkeit [299].
-
-
Strahlentherapie:
-
zusätzliche Bestrahlung in manchen Studien ohne nachweisbaren Überlebensvorteil [288], in anderen Studien Nachweis einer Verbesserung der lokalen Kontrolle nach adjuvanter Bestrahlung bei größeren Tumoren [297]
-
Partikelbestrahlung: Protonen- und Schwerionenbestrahlung mit befriedigendem Erfolg bei ACC der Tränendrüse [300] [301] [302] [303]
-
Brachytherapie: kann Teil des Therapiekonzepts sein, z. B. zur Behandlung von Patienten nach R1-Resektion [304] [305]
-
-
Chemotherapie: Hinweise auf Verbesserung der Prognose durch Intraarterielle zytoreduktive Chemotherapie (IACC) vor (und nach) der Therapie mittels OP und Bestrahlung [289] [290], außerdem Nachweis einer erhöhten Rate an augenerhaltenden Operationen nach neoadjuvanter IACC [306]
-
-
Prognose:
-
hohe Lokalrezidivrate
-
schlechte Langzeitprognose: mediane Überlebensdauer: 5 Jahre, 10-Jahres-Überleben: 20%
-
ACC der Tränendrüse mehrheitlich vom High-grade-Phänotyp mit aggressiverem Verhalten und kürzerem medianem Überleben (2,5 Jahre) [297]
-
häufig Tod durch intrakranielles Tumorwachstum oder Lungenmetastasen
7.2.1.2.1.2 Karzinom im pleomorphen Adenom [252] [261]
-
Definition:
gemäß WHO-Klassifikation der Speicheldrüsentumoren von 2017 Verwendung des Begriffs „Karzinom im pleomorphen Adenom“ nur noch zusammen mit dem histologischen Subtyp [265]
-
Synonyme:
Pleomorphes (Adeno-)Karzinom, maligner Mischtumor (irreführend, da nur der epitheliale (Karzinom), nicht aber der mesenchymale Anteil entartet ist; anders ist dies beim Karzinosarkom („echter“ maligner Mischtumor) welches in der Tränendrüse ebenfalls beschrieben ist [282])
-
Epidemiologie:
-
zweithäufigster bösartiger epithelialer Tränendrüsentumor (4–18%) [279] [291] [307] [308] [309]
-
Auftreten typischerweise in der 6./7. Lebensdekade (etwa 10 Jahre später als das pleomorphe Adenom)
-
Adenokarzinome häufiger bei Männern, adenoidzystische Karzinome häufiger bei Frauen [287]
-
Einteilung:
-
nicht invasiv (intrakapsulär bezogen auf pleomorphes Adenom)
-
minimalinvasiv (< 4–6 mm über die Kapsel)
-
invasiv (> 4–6 mm über die Kapsel) [265]
-
Histologie:
-
zumeist gering differenziertes Adenokarzinom oder undifferenziertes Karzinom, seltener adenoidzystisches Karzinom
-
auch plattenepithelial differenzierte Karzinome und Spindelzellsarkome möglich [235] [261] [308] [310]
-
Klinik:
plötzliches Tumorrezidiv nach unvollständiger Entfernung eines pleomorphen Adenoms oder plötzliches deutliches Wachstum (eventuell mit begleitendem Schmerz) eines seit Jahren bestehenden, beschwerdefreien Tumors
-
Therapie:
-
Tumorresektion (gegebenenfalls radikale Orbitektomie einschließlich knöcherner Anteile, je nach Ausdehnung) mit Entfernung regionaler (Parotis) und zervikaler Lymphknoten (Adenokarzinome können frühzeitig lymphatisch metastasieren)
-
Adjuvante Strahlentherapie im Anschluss als sinnvolle Ergänzung möglich [296] [311]
-
Prognose:
-
meist als eher ungünstig angegeben, allerdings abhängig von zahlreichen Faktoren wie histologischem Subtyp, Invasivität u. a.
-
nicht invasive und minimalinvasive Formen: sehr gute Prognose nach vollständiger chirurgischer Resektion (ohne adjuvante Therapie)
-
invasive Formen: sehr aggressive Tumoren mit schlechter Prognose, aber günstigem prognostischem Effekt der adjuvanten Therapie [312] [313]
-
Todesursachen: intrakranielle Ausbreitung, Fernmetastasen (Lunge, Thoraxwand, Knochen)
7.2.1.2.1.3 Adenokarzinom (not otherwise specified, NOS) [233]
Anmerkung: In der aktuellen Klassifikation der Speicheldrüsentumoren der WHO wurde die Anzahl der Entitäten von 39 in der vorangegangenen Ausgabe auf 33 gesenkt, um die Komplexität zu reduzieren und die Klassifikation übersichtlicher zu gestalten. Dies führte unter anderem dazu, dass mehrere, teils extrem seltene Entitäten zu den „Adenokarzinomen NOS“ zusammengefasst wurden [285]
-
Epidemiologie:
dritthäufigster bösartiger epithelialer Tränendrüsentumor (5–10%) [237] [279]
-
Ätiologie:
de novo oder innerhalb eines pleomorphen Adenoms
-
Pathologie:
-
hochmaligne
-
metastasiert im Vergleich zum adenoidzystischen Karzinom früher
-
frühzeitige lymphatische und hämatogene Metastasierung v. a. in Lunge, Gehirn und Mediastinum [314]
-
Therapie:
-
häufig bei Erstvorstellung so fortgeschritten, dass eine adäquate chirurgische Resektion erschwert bis unmöglich ist
-
Operative Therapie, gegebenenfalls mit Exenteratio orbitae oder kraniofazialer Orbitektomie und regionaler Lymphknotendissektion [291] [315]
-
Her-2-Überexpression in Adenokarzinomen der Tränendrüse beobachtet – Medikamente wie Lapatinib als mögliche therapeutische Option [317]
-
Prognose:
kürzere Überlebenszeit im Vergleich zum adenoidzystischen Karzinom
7.2.1.2.1.4 Mukoepidermoidkarzinom
-
Epidemiologie:
-
vierthäufigster bösartiger epithelialer Tränendrüsentumor (2–3%) [237] [279]
-
bis zum Jahr 2000 weniger als 30 Fallberichte in der Literatur [318]
-
durchschnittliches Erkrankungsalter: 49 Jahre
-
Klink:
typischerweise schmerzlose, langsam wachsende Raumforderung
-
Differenzialdiagnose:
nicht selten Verwechslung mit pleomorphem Adenom [296] [319] [320]
-
Therapie:
-
High-grade-Tumoren: Exenteratio (mit Resektion betroffener Knochen) / Orbitektomie und adjuvante Radiatio
-
Low-grade-Tumoren: Tumorexstirpation (+/− Radiatio)[296] [319]
-
regionale Lymphknotendissektion (Parotidektomie und Neck Dissection), v. a. bei höherem Grading und/oder entsprechender Tumorgröße
-
Prognose [314]:
-
tumorfreies Überleben und Gesamtprognose maßgeblich abhängig vom Grading (Einteilung in Grad I–III anhand histopathologischer Eigenschaften)
-
Thorvaldsson et al. (1970): medianes Follow-up von 12 Jahren bei Mukoepidermoidkarzinomen der großen Speicheldrüsen: Überlebensraten von 100% bzw. 97% bei Patienten mit Grad-1- bzw. Grad-2-Tumoren, aber nur 43% bei Grad-3-Tumoren (high grade) [314] [321]
7.2.1.2.1.5 Duktales Adenokarzinom
-
Epidemiologie:
-
Ätiologie:
Entstehung als Karzinom im pleomorphen Adenom möglich [324]
-
Pathologie:
-
-
Therapieempfehlungen in Anlehnung an das duktale Speicheldrüsenkarzinom
-
chirurgische Resektion, ggf. mit adjuvanter Radio(chemo)therapie
-
Exenteratio orbitae bei > 50% der operierten Patienten
-
primäre Radiochemotherapie bei nicht resektablen Tumoren
-
Chemotherapie im palliativen Setting mit Ansprechraten von 15–50%
-
gegebenenfalls medikamentöse Androgendeprivation in der rezidivierten oder metastasierten Situation, z. B. Bicalutamid bei Androgenrezeptor-positivem Tumor, beziehungsweise z. B. Trastuzumab oder Lapatinib bei Her-2/neu-Expression
-
-
Prognose:
7.2.1.2.1.6 Azinuszellkarzinom [328] [329] [330] [331]
-
Epidemiologie:
-
sehr selten im Bereich der Speicheldrüsen (2–4% der Parotistumoren)
-
noch deutlich seltener im Bereich der Tränendrüse, einzelne Fallberichte in der Literatur
-
Frauen scheinen häufiger betroffen zu sein
-
Manifestationsalter: 6. Lebensdekade
-
-
Diagnostik/Differenzialdiagnose:
uncharakteristisch in der CT-/MRT-Bildgebung, daher hohe Verwechslungsgefahr mit einem pleomorphen Adenom
-
Therapie:
chirurgische Resektion, je nach Ausmaß der Läsion augenerhaltend, ggf. mit Exenteratio oder erweiterter Exenteratio (bei Knocheninfiltration)
7.2.1.2.1.7 Primäres Talgdrüsenkarzinom (engl. Sebaceous Carcinoma) [332] [333] [334] [335]
-
Epidemiologie:
extrem selten, nur einzelne Fallberichte
-
Hypothesen zur Pathogenese:
-
ausgehend von heterotopem Gewebe
-
maligne Transformation und talgdrüsenartige Differenzierung anderer epithelialer Tumoren der Tränendrüse
-
ausgehend von entarteten pluripotenten Zellen
-
-
Pathologie:
hochmaligner Tumor, der oft sehr spät erkannt wird (high-grade)
-
Differenzialdiagnosen:
-
abzugrenzen von sekundärer Invasion (z. B. ausgehend von Oberlidkarzinom) und metastatischer Absiedelung
-
histologisch teilweise schwer abzugrenzen von anderen Entitäten wie Plattenepithelkarzinomen oder Basalzellkarzinomen (40–75% der Talgdrüsenkarzinome der Tränendrüse wurden initial für Plattenepithelkarzinome gehalten, spezielle Anfärbung für Fett kann diese Verwechslung vermeiden)
-
-
Therapie:
-
operative Therapie, ggf. Exenteratio orbitae oder erweiterte Exenteratio
-
Parotidektomie / Neck Dissection abhängig von Ausdehnung (Infiltration der Umgebungsstrukturen, z. B. des Oberlids)
-
adjuvante Strahlentherapie (Lokalrezidivrate nach Resektion 9–36%)
-
alleinige Strahlentherapie im Falle inoperabler Tumoren
-
Partikeltherapie (z. B. Schwerionen, C12) [336]
-
7.2.1.2.1.8 Myoepitheliales Karzinom [337] [338] [339] [340] [341]
-
Epidemiologie:
-
Pathologie:
die histopathologische Diagnostik ist erschwert und es kommt zu Fehlinterpretationen aufgrund der Seltenheit der Erkrankung und des histologischen Polymorphismus [286] [342]
-
Therapie:
-
insgesamt so selten, dass kein bewährtes Therapiekonzept existiert
-
operative Therapie:
-
Strahlentherapie:
-
Anwendung bei myoepithelialen Tumoren der Tränendrüse zumindest in palliativer Form [344], bei Befall der Glandula parotidea oder anderer Lokalisationen im Kopf-Hals-Bereich in etwa der Hälfte der Fälle als adjuvante Therapie nach operativer Resektion [341]
-
primäre Radiotherapie, wenn chirurgische Resektion nicht möglich
-
Stellenwert der adjuvanten Radiatio unklar, Berichte mehrerer Autoren über einen fehlenden Effekt auf die Lokalrezidivrate [345]
-
-
Chemotherapeutika teilweise als wirksam beschrieben, jedoch ohne Vorliegen systematischer Daten
-
-
Prognose:
-
High-grade-Malignom mit schlechter Prognose und hoher Rezidivrate [286] [341]
-
myoepitheliale Karzinome der Kopf-Hals-Region: 5-Jahres-Überlebensrate 32% [341]
-
Fernmetastasierungsrate von 35% (v. a. pulmonale Metastasen) [341]
-
Fernmetastasen mit Primarius in der Tränendrüse kaum beschrieben (CAVE: geringe Fallzahl!) [342]
-
7.2.1.2.1.9 Plattenepithelkarzinom
-
Epidemiologie:
nach Review-Artikel von Weis et al. (2009) bis zu diesem Zeitpunkt nur acht Fallberichte in der Literatur [286]
-
Ätiologie/Pathogenese:
Entstehung mehrerer der wenigen Fälle auf dem Boden einer epithelial ausgekleideten Zyste der Tränendrüse (Dacryops) [346] [347]
-
Therapie:
chirurgische Resektion (ggf. Exenteratio orbitae) und adjuvante Strahlen(chemo)therapie [346] [347] [348]
7.2.1.2.1.10 Onkozytisches Karzinom [349] [350]
-
Epidemiologie:
nach Kalantzis et al. (2013) bis zu diesem Zeitpunkt nur vier Fälle mit Befall der Tränendrüse in der Literatur [350]
-
Pathologie:
High-grade-Tumor mit infiltrativem Wachstumsmuster, Neigung zu Lokalrezidiven und Fernmetastasen
-
Therapie:
radikale Resektion (ggf. Exenteratio orbitae) gefolgt von adjuvanter Radio(chemo)therapie
7.2.1.2.1.11 Polymorphes Adenokarzinom [265] [351]
-
frühere Bezeichnung (bis 2017):
Polymorphes Low-grade-Adenokarzinom
-
Epidemiologie:
-
nach Selva et. al (2004) bis zu diesem Zeitpunkt nur vier Fälle mit Befall der Tränendrüse in der Literatur [351]
-
Kommt fast ausschließlich in kleinen Speicheldrüsen vor, vor allem am Gaumen (w : m = 2 : 1)
-
-
Differenzialdiagnose:
nicht selten Verwechslung mit Adenoidzystischem Karzinom
-
Pathologie:
-
neben infiltrativem Wachstum auch perineurale Invasion
-
Metastasen in regionalen Lymphknoten relativ selten (6,5–10%), Fernmetastasen extrem selten
-
-
Therapie:
-
chirurgische Resektion (ggf. Exenteratio orbitae); aufgrund der relativ günstigen Prognose gemäß Selva et al. (2004) möglichst augenerhaltende Resektion erwägen
-
Notwendigkeit einer adjuvanten Radiatio fraglich [351]
-
-
Prognose:
-
deutlich besser als bei anderen Adenokarzinomen der Speicheldrüse („overall survival“ ≥ 95%)
-
bei Befall der Speicheldrüsen Lokalrezidive in 5–33% der Fälle (in der Regel durch erneute Resektion gut beherrschbar)
-
7.2.1.2.1.12 Sekretorisches Karzinom[265] [352] [353]
-
Definition:
-
neue Entität, erstmals 2010 im Kopf-Hals-Bereich als mammary analogue secretory carcinoma beschrieben, von der WHO kurz „Sekretorisches Karzinom“ genannt
-
davor Zuordnung zu anderen Entitäten trotz nicht immer typischer histopathologischer Eigenschaften (z. B. „granule poor acinic cell carcinoma“)
-
Pathologie:
-
dem sekretorischen Karzinom der Brust ähnlich, ebenfalls Nachweis des spezifischen ETV6-NTRAK3-Fusionsgens
-
Vorkommen im Kopf-Hals-Bereich, v. a. in der Glandula parotidea (70%), daneben in Wangen-, Lippen- und Gaumenschleimhaut, selten in den Glandulae submandibularis oder sublingualis
-
regionale Lymphknotenmetastasen in bis zu 25% der Fälle
-
Therapie:
-
chirurgische Resektion
-
Tropomyosin-Rezeptor-Kinase-(TRK-) Inhibitoren (Larotrectinib, Entrectinib) als künftige Therapieoption für Tumoren mit NTRK-Fusion
-
Prognose:
insgesamt wenig aggressiver Tumor mit relativ günstiger Prognose („survival exceeding: 95%“)
7.2.1.2.2 Bösartige Tumoren der Tränendrüse nicht epithelialen Ursprungs
Bösartige Tumoren der Tränendrüse nicht epithelialen Ursprungs werden in [Tab. 7] zusammengefasst. Lymphome der Tränendrüse werden in Kapitel 7.2.1.2.3 gesondert behandelt.
|
Neoplasie |
Charakteristika |
|---|---|
|
Synovialsarkom [354] |
▪ Epidemiologie:
▪ Pathologie:
|
|
▪ Ätiologie:
▪ Therapie:
|
|
|
maligner peripherer Nervenscheidentumor [357] [358] (Vermeidung der synomym verwendeten Begriffe „neurogenes Sarkom“, „Neurofibrosarkom“ und „malignes Schwannom“) |
▪ Epidemiologie:
▪ Risikofaktoren:
▪ Pathogenese:
▪ Pathologie:
▪ Diagnostik:
▪ Therapie:
▪ Prognose:
|
|
▪ Definition:
▪ Epidemiologie:
▪ Ätiologie/Pathogenese:
▪ Therapie:
|
7.2.1.2.3 Maligne lymphoproliferative Erkrankungen – Lymphome der Tränendrüse [233] [237] [365] [366]
-
Definition:
zu den okulären Adnexlymphomen (OAL) gehörend
-
Epidemiologie:
-
Lymphome stellen die häufigsten orbitalen Malignome dar [365]
-
37% aller malignen Tumoren der Tränendrüse sind Lymphome [366]
-
vorwiegend ältere Menschen betroffen; Altersmedian: 60(+) Jahre
-
w : m = 2,4 : 1
-
Einteilung: in absteigender Häufigkeit findet man in der Tränendrüse:
-
Extranodales Marginalzonenlymphom (EMZL)
-
Follikuläres Lymphom (FL)
-
Diffus-großzelliges B-Zell-Lymphom (DLBCL)
-
Ätiologie/Pathogenese:
in seltenen Fällen Entstehung auf dem Boden chronischer entzündlicher Erkrankungen (Sjögren-Syndrom, IgG4-assoziierte Erkrankungen, reaktive lymphatische Hyperplasie)
-
Klinik:
-
Hauptsymptome: Raumforderung, Bulbusdislokation, Doppelbilder
-
Schmerzen nur selten (im Rahmen knöcherner Infiltration bei hochmalignen Formen)
-
eventuell lachsfarbener Tumor im Bereich der angrenzenden Konjuktiva [233]
-
typischerweise Befall des orbitalen und palpebralen Anteils der Drüse [233]
-
Diagnostik:
-
bildgebende Verfahren:
-
CT oder MRT: glatte und homogene Raumforderung meist ohne Infiltration des umgebenden Gewebes (aber knöcherne Arrosion bei hochaggressiven Formen möglich [365])
-
sichere Unterscheidung zwischen reaktiver lymphatischer Hyperplasie, Lymphom und unspezifischer Entzündung der Tränendrüse klinisch und bildmorphologisch nicht möglich → Biopsie erforderlich (inzisionale Biopsie ist der Feinnadelbiopsie vorzuziehen!) [233]
-
weitere Diagnostik (zur Abklärung einer systemischen Beteiligung):
-
Knochenmarkbiopsie, Ganzkörper-CT/-MRT oder PET
-
Ann-Arbor-Klassifikation und TNM-Staging werden verwendet
-
Therapie:
-
Strahlentherapie, insbesondere bei lokalisierter Form (Ann-Arbor-Stadium IE)
-
Polychemotherapie, insbesondere bei systemischer Ausbreitung (Stadien IIE–IVE), bei DLBCL auch im Frühstadium [369]
-
Antikörpertherapie, z. B. Anti-CD-20-Antikörper Rituximab
-
gegebenenfalls Stammzelltransplantation
-
chirurgische Therapie: alleinige chirurgische Therapie in der Regel nicht gerechtfertigt [233]
-
Prognose:
-
etwa 80% befinden sich zum Zeitpunkt der Erstdiagnose in einem lokalisierten Stadium (IE), etwa 20% zeigen im Rahmen des Stagings eine systemische Beteiligung (Stadium IIE–IVE) [233]
-
Patienten mit Stadium IE bei Erstdiagnose: sekundäre Generalisierung im Verlauf in 22–42% der Fälle [233]
-
Rasmussen et al. (2011): 5-Jahres-Überlebensrate von 70% bei 27 Patienten mit Lymphom der Tränendrüse [366]
7.2.1.2.3.1 Extranodales Marginalzonen-B-Zell-Lymphom
-
Epidemiologie:
mit 40% aller Lymphome häufigster Subtyp in der Tränendrüse [366]
-
Therapie:
-
Early Stage (Stadium IE/IIE, kein systemischer Befall): Strahlentherapie (ca. 30–40 Gy) [370]
-
bei systemischem Befall oder hoher Tumorlast: medikamentöse Kombinationstherapie, z. B. Rituximab+ Cyclophosphamid+ Hydroxydaunorubicin+ Vincristin+ Prednison (R-CHOP) → 3-Jahres-progressionsfreies Überleben in 70–80%
-
Rituximab wird zwei Jahre als Erhaltungsdosis weitergegeben
-
-
Prognose:
Low-grade-Lymphom; 5-Jahres-Überlebensrate von 75% [366], bei Speicheldrüsen wohl besser (95–100%)
7.2.1.2.3.2 Follikuläres Lymphom
-
Epidemiologie:
mit etwa 28% aller Lymphome zweithäufigster Subtyp in der Tränendrüse [365] [366]
-
Therapie:
-
Early Stage (Stadium IE/IIE): Strahlentherapie → 5-Jahres-Überlebensrate bei 100% [366]
-
bei systemischem Befall oder hoher Tumorlast: medikamentöse Kombinationstherapie, z. B. Rituximab+ Cyclophosphamid+ Hydroxydaunorubicin+ Vincristin+ Prednison (R-CHOP) → 3-Jahres-progressionsfreies Überleben in 70–80%
-
Rituximab wird zwei Jahre als Erhaltungsdosis weitergegeben
-
-
Prognose:
Low-grade-Lymphom mit günstiger Prognose (5-Jahres-Überlebensrate bei 80–90%)
7.2.1.2.3.3 Diffus-großzelliges B-Zell-Lymphom
-
Epidemiologie:
mit 15% aller Lymphome dritthäufigster Subtyp in der Tränendrüse [365] [366]
-
Pathologie:
destruktives Wachstum in benachbarte Strukturen (Ethmoid, intrakraniell…) bei 6 von 27 Patienten beobachtet (Ferry et al. (2007) [365])
-
Therapie:
-
bereits bei Stadium I (Befall nur der Tränendrüse): Rituximab+ Cyclophosphamid+ Hydroxydaunorubicin+ Vincristin+ Prednison (R-CHOP)
-
bei Patienten <60 Jahren mit hohem Risikoprofil (IPI-Score ≥2 oder erhöhte LDH) zusätzlich Etoposid (R-CHOEP)
-
bei Stadium IE und Fehlen von B-Symptomen zusätzlich Strahlentherapie
-
-
Prognose:
High-grade-Lymphom; 5-Jahres-Überlebensrate: 55 –73%
7.2.1.2.3.4 Mantelzelllymphom
-
Therapie:
-
Primärtherapie: R-CHOP
-
bei Patienten < 65 Jahren zusätzlich Cytarabin und Stammzelltransplantation
-
-
Prognose: ungünstig
Anmerkung: Die WHO-Klassifikation lymphoider Neoplasien umfasst mehr als 80 Lymphomklassen. Diese werden in drei Gruppen unterteilt: B-Zell-Neoplasien, T-Zell- und NK-Zell-Neoplasien sowie Hodgkin-Lymphome. Sowohl Lymphome als auch lymphoide Leukämien sind enthalten. An dieser Stelle seien nur die wichtigsten Vertreter genannt [371]. Die Häufigkeitsverteilung der Subtypen in der Tränendrüse entspricht der in den Speicheldrüsen [366].
7.2.1.3 Metastasen in der Tränendrüse
Epidemiologie:
-
in 80% der Fälle ist bei Erstdiagnose der orbitalen Raumforderung eine maligne Grunderkrankung bekannt [233]
-
Vorliegen orbitaler Metastasen (meist einseitig) bei geschätzt 2–3% aller Patienten mit Malignomen[233]
7.2.2 Neoplasien der Orbita (ohne Neoplasien der Tränendrüse)
7.2.2.1 Gutartige Neoplasien
7.2.2.1.1 Gutartige Neoplasien epithelialen Ursprungs
Siehe Kapitel 6.2.1.1.1 „Gutartige epitheliale Neoplasien der Tränendrüse“
7.2.2.1.2 Gutartige Neoplasien nicht epithelialen Ursprungs
Vielfältiger als die Tumoren epithelialen Ursprungs der Orbita sind die Tumoren nicht epithelialen Ursprungs.
7.2.2.1.2.1 Fibro-ossäre Prozesse der Orbita
Es existieren eine Reihe von fibro-ossären Läsionen, beispielsweise die aneurysmatische Knochenzyste, das Riesenzellgranulom oder Cholesterolgranulome, die in der periorbitalen Region auftreten und sekundär auch zu einer Affektion der orbitalen Weichteile führen können. In der Literatur werden sie jedoch eher als reaktive knöcherne Läsionen bezeichnet und zumeist zu den knöchernen Tumoren der Orbita gezählt.
7.2.2.1.2.1.1 Fibrom, ossifizierendes Fibrom, Fibromatose [201] [373]
Fibrome
-
Epidemiologie:
-
sehr selten in der Orbita
-
7 Fibrome bei 2210 orbitalen Tumoren aus vier Fallserien [373] [374] [375]
-
Auftreten meist in der Adoleszenz und im frühen Erwachsenenalter
-
Pathologie:
meist ausgehend von der kranialen Periorbita oder dem Muskelkonus
Ossifizierende Fibrome [376]
-
Epidemiologie:
Auftreten v. a. bei jüngeren Patienten
-
Pathologie:
-
langsam wachsende, gutartige Tumoren, destruktives Wachstum und Rezidive nach unzureichender Resektion aber möglich
-
Lokalisation auch in den Nasennebenhöhlen oder Kieferknochen
-
Diagnostik:
radiologische Abgrenzung von fibrösen Dysplasien notwendig
Fibromatosen
-
können von intermediärem Charakter sein
-
Therapie:
in der Regel chirurgische Resektion
7.2.2.1.2.1.2 Aneurysmatische Knochenzyste (AKZ)
-
Definition:
lokal expansive, osteolytische Läsion, bestehend aus blutgefüllten Hohlräumen mit umgebendem reaktivem, fibro-ossärem Gewebe
-
Lokalisation:
-
Epidemiologie:
bevorzugtes Auftreten im Kindes- und Jugendalter bis zum 20. Lebensjahr [381]
-
Ätiologie und Pathogenese:
-
Assoziation zu anderen knöchernen Läsionen (fibröse Dysplasie, Chondroblastom, Osteoblastom, Riesenzelltumoren), die dem Auftreten von AKZ vorausgehen und für ihre Entwicklung als reaktives Phänomen prädisponierend sein können [382] [383]
-
als kausale Ursache lange Zeit in der Diskussion: fokale hämodynamische Veränderungen mit sekundärer venöser Hypertonie bzw. vorbestehende Traumata [381]
-
aktuelle onkogenetische Studienergebnisse: klonale neoplastische Läsion mit spezifischem genetischem Rearrangement (USP6-Gen-Hochregulation in Spindelzellen) [384] → dadurch zukünftig Differenzierung zu ähnlichen Läsionen ohne USP-Rearrangement (z. B. Osteosarkome, Chondroblastome, Riesenzelltumore) möglich
-
Klinik:
-
initial indolente Schwellung oder Protrusio bulbi
-
häufig langsames Wachstum über Wochen bis Monate, dann Episoden mit sehr raschem Wachstum und schmerzhaftem Exophthalmus, Ptosis, Diplopie und weiteren ophthalmologischen Symptomen [378]
-
Diagnostik:
-
CT und MRT (charakteristische Veränderungen; [Abb. 16a+b]): polylobulierte Raumforderung mit multiplen zystischen Arealen, durch Bindegewebssepten unterteilt; weiterhin solide Anteile, Ausdünnung der umgebenden Kortikalis, Flüssigkeitsspiegel in den zystischen Arealen, bedingt durch nicht koaguliertes Blut; deutliche KM-Aufnahme; Dura wird bei intrakranieller Ausbreitung respektiert [378] [385]
-
Histologie: von endothelähnlichen Zellen ausgekleidete zystische Hohlräume; solider Anteil, bestehend aus spindelförmigen Fibroblasten mit vereinzelten vielkernigen Riesenzellen, hämosiderinbeladenen Makrophagen, Granulationsgewebe und extravasalen Erythrozyten; Septen häufig mit neu gebildeten Knochen; keine atypischen Mitosen oder andere histologische Malignitätskriterien

-
Differenzialdiagnosen:
-
andere Knochentumoren (z. B. Osteoblastome, Osteoklastome, braune Tumoren)
-
reparatives Riesenzellgranulom und Riesenzelltumoren
-
Abklärung durch pathologische Referenzzentren erforderlich [378]
-
Therapie:
-
erste Wahl: chirurgische Exzision und (je nach Ausdehnung und lokaler Destruktion) Rekonstruktion des knöchernen Defekts
-
in ausgedehnten Fällen: intraläsionale Kürettage und partielle Entfernung mit adjuvanter Strahlentherapie
-
bei hohem intraoperativem Blutungsrisiko: präoperative Embolisation oder stereotaktische Radiochirurgie sinnvoll
-
Prognose:
-
Rezidivrate 10–30%
-
maligne Transformation möglich (extrem selten) [384]
7.2.2.1.2.1.3 Riesenzellgranulom [387] [388] [389] [390] [391]
-
Definition:
seltene, reaktive, benigne Raumforderung mit lokal aggressivem Wachstum
-
Epidemiologie:
meist bei Frauen in der 1.–3. Lebensdekade [392]
-
Lokalisation:
-
Epiphysen der Röhrenknochen (> 90% der Fälle)
-
Schädel (ca. 2% der Fälle): meist Mandibula und Maxilla, seltener Sphenoid und Felsenbein, ethmoidal oder orbital [387] [391]
-
Pathogenese:
-
noch unklar
-
Inflammation, Trauma und intraossäre Hämorrhagien als Ursache möglich
-
aufgrund Assoziation mit anderen Syndromen wie Noonan-Syndrom, Neurofibromatose Typ I und Cherubismus auch bislang unentdeckte Chromosomenanomalie denkbar [393]
-
Klinik:
-
Symptome meist durch raumfordernden Effekt: Schwellung, Deformität, gelegentlich Schmerzen
-
Exophthalmus, Visuseinschränkungen, Diplopie als klinische Erstsymptome
-
Diagnostik:
-
CT: meist scharf begrenzter, osteolytischer Prozess mit expansivem Charakter, jedoch sehr variabel und unspezifisch
-
Histopathologie: charakteristische Ansammlung mehrkerniger, großer, Osteoklasten-ähnlicher Riesenzellen mit einer Tendenz zur Clusterbildung um hämorrhagische Foci
-
Differenzialdiagnosen:
-
Osteoklastom, Osteosarkom, eosinophiles Granulom, aneursmatische Knochenzyste, „braune Tumoren“ bei Hyperparathyreoidismus, nicht ossifizierendes Fibrom, Langerhans-Zell-Histiozytose (radiologische Abgrenzung teilweise schwierig)
-
Abgrenzung zu Riesenzelltumoren in der Literatur kontrovers diskutiert [394]: endgültige Diagnosestellung des Riesenzellgranuloms der Orbita in den beschriebenen Fällen erst durch Biopsie oder chirurgische Exzision möglich
-
Therapie:
-
erste Wahl: vollständige Resektion
-
im Bereich der Schädelknochen nicht immer radikal durchführbar
-
Radiatio: bei Strahlenresistenz und möglicher maligner Zellentartung kontrovers diskutiert, jedoch in einigen Quellen nach partieller Resektion empfohlen
-
Medikamente zur adjuvanten Therapie: Kortikosteroide, Calcitonin, Bisphosphonate, Imatinib und Interferon-α [393] [395]
-
Denosumab (monoklonaler Antikörper): Neutralisierung durch Bindung an den von den Tumorzellen produzierten RANK-Liganden bei inoperablen oder lokal fortgeschrittenen Fällen [391]
-
Prognose:
Rezidive in 10–50% der Fälle
7.2.2.1.2.1.4 Cholesteringranulom
-
Definition:
seltene, gutartige Läsion, durch Fremdkörperreaktion auf Cholesterinkristalle entstehend
-
Epidemiologie:
Auftreten typischerweise bei Männern mittleren Alters [396]
-
Ätiologie und Pathogenese:
-
Entstehung durch traumatische Genese oder intrinsische Anomalie der Diploe vermutet
-
inflammatorische Reaktion gegen Abbauprodukte von Blutbestandteilen bzw. bei hypoxischem Gewebsuntergang
-
granulomatöse Entzündungsreaktion auf Cholesterin mit darauffolgender Arrosion der umgebenden Knochen [397] [398]
-
-
Lokalisation:
-
meist im Schläfenbein
-
auch im Bereich des Gesichtsschädels möglich
-
selten orbitale Lokalisation, dann typischerweise im Bereich des Stirnbeins oberhalb der Fossa glandulae lacrimalis (superotemporal oder superior), da hier schwächste Verankerung der Periorbita am Knochen mit häufigem Auftreten von Traumata [399] [400] [401]
→ bei unklarer Läsion der Orbita in Zusammenschau von Bildgebung, Lokalisation und häufig positiver Traumaanamnese auch Cholesteringranulom in Erwägung ziehen! [400]
-
intrakonale Lokalisation und Auftreten am Orbitaboden in einigen Fällen beschrieben [401]
-
Klinik:
-
Exophthalmus, Diplopie, Visuseinschränkungen
-
seltener periorbitale Schmerzen oder Ptosis
-
Diagnostik:
-
CT: osteolytische Läsionen der Diploe des Stirnbeins mit Arrosion der angrenzenden Kortikalis und ggf. Expansion in den Extraduralraum
-
MRT: Unterscheidung zu anderen Weichteiltumoren möglich: typischerweise hohe Signalintensitäten in T1- UND T2-gewichteten Sequenzen aufgrund des Lipidgehalts [402]
-
Histologie: Cholesterolkristalle, umgeben von vielkernigen Riesenzellen; Unterscheidung von Epidermoiden und Dermoidzysten durch Abwesenheit von epithelialen Elementen; Abgrenzung von der aneurysmatischen Knochenzyste oder von Riesenzelltumoren durch die prominente xanthomatöse Komponente
-
Differenzialdiagnosen:
-
klinisch und CT-morphologisch Abgrenzung zu einer Dermoid- oder Epidermoidzyste erschwert
-
weitere: Neoplasien der Tränendrüse, Metastasen, aneurysmatische Knochenzysten oder Chondrome [61]
-
Therapie:
-
erste Wahl: Drainage des Inhalts der Läsion und metikulöse Entfernung der Zystenwand sowie des umgebenden Knochens → Minimierung des Rezidivrisikos
-
verschiedene Techniken, abhängig von Größe und Lokalisation der Läsion [400] [401] [403]
7.2.2.1.2.1.5 Osteom [201]
-
Epidemiologie:
häufigster vom Orbitaknochen ausgehender Tumor (häufig auch ausgehend von den Nasennebenhöhlen)
-
Diagnostik:
im CT knochendichte, gut begrenzte Raumforderung (häufig Zufallsbefund)
-
Therapie:
chirurgische Resektion, nur bei symptomatischen/progredienten Prozessen
7.2.2.1.2.1.6 Chondrom [201] [404]
Anmerkung: Trochlea ist einzige kartilaginäre Struktur der Orbita, Keilbeinknochen hat knorpelige Vorläuferstrukturen.
-
Epidemiologie:
-
Garrity und Henderson (2007) [405] bzw. Shields et al. (2004) [279] : nur je ein Fall unter 1373 bzw. 627 mesenchymalen Orbitatumoren
-
Rootman et al. (2004): ein Fall unter 62 primären Tumoren des Orbitaknochens in einem Zeitraum von 24 Jahren [405] [406] [407]
-
im Bereich des Gesichtsschädelknochens vor allem bei Jugendlichen und jungen Erwachsenen
-
-
Klinik:
-
häufig asymptomatisch
-
bei Größenzunahme Ptose/Proptose
-
-
Lokalisation:
nasaler oberer Quadrant
-
Pathologie:
-
histopathologische Abgrenzung von Low-grade-Chondrosarkomen schwierig
-
teilweise aggressives Verhalten chondrogener Tumoren des Gesichtsskeletts (Diskrepanz zwischen histologischem Bild und biologischem Verhalten)
-
-
Therapie:
-
Resektion, wenn möglich mit Sicherheitsabstand
-
Langzeit-Follow-up empfohlen [404]
-
7.2.2.1.2.2 Neurinom [201] [408]
-
Synonyme:
Schwannom, Schwann-Zell-Tumor
-
Epidemiologie:
-
0,7–2,3% der orbitalen Tumoren
-
in der Regel singulär
-
in bis zu 18% der Fälle Assoziation mit Neurofibromatose
-
-
Pathologie:
gutartige Tumoren peripherer Nerven
-
Therapie:
chirurgische Resektion, insbesondere bei symptomatischen Prozessen
7.2.2.1.2.3 Neurofibrom [201] [409]
Plexiformes Neurofibrom
-
Epidemiologie:
-
häufigster Subtyp in der Orbita
-
oft kongenital oder im frühen Kindesalter
-
Neurofibromatose Typ I sehr häufig zugrunde liegend
-
Pathologie:
-
multinoduläre Raumforderungen mit infiltrativem Verhalten (Lid, Schädelbasis, Periorbita) und relativ starker Vaskularisation
-
Entartungsrate (zum malignen peripheren Nervenscheidentumor): 2–4%
-
Klinik:
schwere Beeinträchtigung des Visus typisch
-
Therapie:
-
chirurgische Resektion
-
durch unregelmäßige Begrenzung deutlich erschwert, oft multiple Eingriffe bis hin zur Exenteratio
Lokalisiertes Neurofibrom [409] , [ 410]
-
Epidemiologie:
-
selten mit Neurofibromatose assoziiert
-
Manifestationsgipfel: 3.–5. Lebensdekade
-
Lokalisation und Pathologie:
typischerweise kranial in der Orbita liegend (Nervus frontalis), weniger vaskularisiert, besser bekapselt
-
Therapie:
chirurgische Resektion
-
Pathologie:
im Unterschied zum Schwannom zusätzlich Fibroblasten und perineurale Zellen, höherer Gehalt an bindegewebiger Substanz, nicht (gut) bekapselt, operativ nicht vom Nerven trennbar (dieser muss daher geopfert werden)
7.2.2.1.2.4 Meningeom [201] [411]
-
Epidemiologie:
-
nach Schätzungen etwa 4–8% aller orbitalen Neoplasien
-
Manifestationsalter: typischerweise etwa um das 45. Lebensjahr, Optikusscheidenmeningeome bei jüngeren Patienten
-
Frauen deutlich häufiger betroffen als Männer
-
-
Risikofaktoren:
-
stattgehabte Strahlentherapie im Kopfbereich
-
Neurofibromatose Typ 2
-
-
typische Lokalisationen:
-
laterale Orbitawand, Orbitadach („primäre ektope Meningeome“, nur Einzelfälle)
-
Optikusscheidenmeningeom (häufigster Optikusscheidentumor), in 90% bei Diagnosestellung bereits Wachstum durch den Optikuskanal Richtung Chiasma [412]
-
Einwachsen von Meningeomen des Sinus cavernosus oder des medialen Keilbeinflügels (Optikuskompression!)
-
-
Pathologie:
orbitale Meningeome in der Regel Grad-1-Meningeome gemäß WHO-Grading für Meningeome (Grad 1: benigne, Grad 2: anaplastisch, Grad 3: maligne) [413]
-
Klinik:
führende Symptome sind
-
allmähliche, schmerzlose Visusminderung (Leitsymptom)
-
Gesichtsfeldausfälle
-
transiente Obskurationen
-
Protrusio oder Motilitätsstörungen (mit Doppelbildern) oft erst im fortgeschrittenen Stadium
-
-
Therapie [411] [412] [414] [415]:
-
bei kleinen und/oder wenig beeinträchtigenden Befunden „watch and wait“ mit zunächst halbjährlichen MRT-Kontrollen
-
Therapieindikationen [412]: progrediente Visusminderung, Visusabfall auf < 0,4, Zunahme der Gesichtsfeldausfälle, Bedrohung des Chiasma
-
Optikusscheidenmeningeom:
-
-
Therapie der ersten Wahl: fraktionierte Strahlentherapie (50–55 Gy) wohl mit bestem funktionellem Outcome (Visus)
-
chirurgische Therapie v. a. bei Ausdehnung nach intrakraniell oder bei Erblindung des Auges und entstellender Protrusion
-
bei Wachstum im Canalis opticus gegebenenfalls Optikus-Dekompression
-
chirurgische Resektion (ggf. prä-OP-Embolisation), eventuell mit adjuvanter Strahlentherapie
-
7.2.2.1.2.5 Optikusgliom [201] [412]
-
Definition:
Sonderform des pilozytischen Astrozytoms (juveniles pilozytisches Astrozytom, WHO Grad I) [412]
-
Epidemiologie:
-
Manifestation in 90% der Fälle vor dem 20. Lebensjahr, in 50% der Fälle vor dem 5. Lebensjahr [412]
-
Neurofibromatose Typ I kann zu Grunde liegen und sollte ausgeschlossen werden
-
-
Differenzialdiagnose:
Optikusschwannom [416] (kommt, wenn auch extrem selten (< 10 Fälle beschrieben), vor, obwohl Myelinscheide des Nervus opticus keine Schwannzellen enthält, geht am ehesten von kleinen Sympathikusfasern aus)
-
Klinik:
-
bilaterale Lokalisation möglich (v. a. bei Neurofibromatose)
-
langsames Wachstum
-
Exophthalmus
-
Schielstellung des betroffenen Auges
-
Visusminderung und Gesichtsfeldeinschränkungen
-
schubweise Zunahme der Beschwerden bei Einblutung
-
Nystagmus
-
endokrinologische Begleiterscheinungen möglich (Diabetes insipidus, Adipositas, Pubertas präcox u. a.)
-
-
-
kontroverse Meinungen, da Spontanverlauf nicht vorhersehbar, spontane Remissionen möglich und alle Verfahren mit teils erheblichen NebenwirkungenNähe zum Canalis opticus entscheidend für den VerlaufVerlaufskontrollen mittels MRT und Visus-/Gesichtsfeldprüfungen (entscheidender für Therapieindikation als radiologische Größenzunahme) bei neu diagnostizierten Gliomen(Teil)resektion v.a. bei großen, fortschreitenden intraorbitalen Gliomen mit Erblindung, Wachstum Richtung Chiasma oder mit entstellendem Exophthalmuseventuell Durchtrennung des Nervus opticus vor dem Chiasma, um eine Ausbreitung auf die Gegenseite zu verhindernfraktionierte Strahlentherapie früher Therapie der Wahl, aber im Kindesalter, v.a. bei unter 7-jährigen, mit Risiko für neurologisch-kognitive Defizite und für spätere sekundäre Tumoren verbunden (insbesondere bei Neurofibromatose-Patienten), daher heutzutage v.a. bei fortschreitenden, chiasmanahen Tumoren älterer Kinder oder Erwachsener indiziertChemotherapie zur Verkleinerung des Tumors, Wachstumsverlangsamung und zum Hinauszögern anderer Therapien möglich. Erstlinientherapie bei Sehverlust / Tumoren mit Handlungsbedarf besonders bei kleinen Kindern. Substanzen z. B. Carboplatin kombiniert mit Vincristin.
-
-
Fallbeispiel 3
-
Anamnese
-
18-jährige Patientin
-
zunehmender Exophthalmus seit etwa vier Monaten und Visusminderung seit fünf Tagen links
-
Diagnostik:
-
Augenärztliche Untersuchung
-
Visus: rechts 1,25, links 0,6
-
Hertel-Exophthalmometrie: rechts 20, links 24 mm
-
Stauungspapille links
-
MRT ([Abb. 17])

-
Verdacht auf 28 x 17 mm großes Optikusgliom
-
kein Wachstum nach intracerebral
-
Prozedere:
-
transnasale, endoskopische mediale Orbitadekompression mit Biopsie
-
Histopathologie: Optikusgliom
-
Definitive fraktionierte stereotaktische Strahlentherapie (50,4 Gy)
-
Verlauf:
-
zunächst schwankender, aber noch guter Visus (um 0,8–0,9)
-
etwa sechs Monate nach Bestrahlung ophthalmologisch beginnende Optikusatrophie, Visus links deutlich verschlechtert (0,25), zunehmendes Papillenödem, sekundärer Strabismus divergens, kosmetisch subjektiv störender progredienter Exophthalmus ([Abb. 18]) und Lidschlussinsuffizienz
-
weitere Verschlechterung in den kommenden drei Monaten (Visus 0,1, Hertel-Exophthalmometrie links 27 mm, zunehmender Leidensdruck, Funduskopie-Befund ([Abb. 19])
-
Enukleatio bulbi und Tumorresektion, Implantation Guthoff-Plombe, später Prothese
-
humangenetische Abklärung zum Ausschluss Neurofibromatose


7.2.2.1.2.6 Myxom [272] [273] [274] [275] [276]
-
Definition:
Neoplasie mesenchymalen/bindegewebigen Ursprungs
-
Epidemiologie:
-
im Bereich der Orbita wenige Fallberichte in der Literatur
-
Männer und Frauen gleichermaßen betroffen
-
-
Pathologie:
-
histologisch zellarm und gefäßarm mit reichlich myxoider (schleimähnlicher) hyaluronsäurereicher Matrix
-
andere Lokalisationen: v. a. Herz, daneben Knochen, Haut, Skelettmuskel, Urogenitaltrakt
-
-
Diagnostik:
-
Assoziation orbitaler Myxome mit Carney-Syndrom (Familiäres Myxom-Syndrom), Mazabraud-Syndrom, McCune-Albright-Syndrom
-
bei Patienten mit vermehrten Leberflecken, einer kardialen Vorgeschichte und einer orbitalen Raumforderung an Myxom denken
-
in der CT gut umschriebene, ovaläre, isodense Raumforderung
-
-
Therapie:
-
vollständige chirurgische Resektion (dann exzellente Prognose)
-
Strahlentherapie ohne relevanten Effekt
-
Weitere Beispiele für gutartige orbitale Neoplasien nicht epithelialen Ursprungs bietet [Tab. 8].
|
Neoplasie |
Charakteristika |
|---|---|
|
Granularzelltumor [420] (Abrikossoff-Tumor) |
▪ Definition:
▪ Epidemiologie:
▪ Pathologie:
▪ Therapie der Wahl:
|
|
▪ Pathologie:
▪ Therapie:
▪ Prognose:
|
|
|
Ganglioneurom [423] [424] (gutartige Form der neuroblastischen Tumoren) |
▪ Epidemiologie:
▪ Pathologie:
▪ Pathogenese:
▪ Therapie:
|
|
Ganglioneuroblastom [423] (intermediäre Form der neuroblastischen Tumoren) |
▪ Pathologie:
|
|
▪ Epidemiologie:
▪ Therapie:
▪ Prognose:
|
|
|
▪ Lokalisation:
▪ Therapie:
|
|
|
▪ Epidemiologie:
▪ Therapie:
|
|
|
pigmentierter neuroektodermaler Tumor [431] [432] [433] (engl. melanotic neuroectodermal tumor of infancy (MNTI), veraltete Synonyme: pigmentiertes Ameloblastom, Retinalanlagetumor, melanotisches Progonom, melanotisches Adamantinom) |
▪ Epidemiologie:
▪ Pathologie:
▪ Therapie:
▪ Prognose:
|
7.2.2.2 Bösartige Neoplasien
7.2.2.2.1 Bösartige Neoplasien epithelialen Ursprungs
Siehe Kapitel 7.2.2.2.3 „Bösartige Tumoren der Tränendrüse epithelialen Ursprungs“
7.2.2.2.2 Bösartige Neoplasien nicht epithelialen Ursprungs
Anmerkung: Lymphome der Orbita werden in Kapitel 6.2.2.2.3 gesondert behandelt.
7.2.2.2.2.1 Rhabdomyosarkom [434]
-
Epidemiologie:
-
häufigstes orbitales Malignom bei Kindern
-
embryonaler Subtyp: häufigster Typ, ca. 60–70% aller kindlichen Rhabdomyosarkome
-
alveolärer Subtyp: 30% aller kindlichen Rhabdomyosarkome, in 80% mit spezieller Gentranslokation/-fusion (PAX3 auf Chromosom 2 oder PAX7 auf Chromosom 1 mit FOXO1 auf Chromosom 13) assoziiert
-
-
Klinik:
-
rasch progredienter Exophthalmus
-
Lidschwellung/-rötung
-
Ptosis, evtl. palpabler Tumor
-
-
Diagnose:
Biopsie
-
Therapie:
-
Standard: Chemotherapie + Radiotherapie
-
Exenteratio sollte nicht primär erfolgen, erst bei Nachweis eines Residuums/Rezidivs in der Biopsie nach Chemotherapie + Radiotherapie erwägen
-
-
Prognose:
-
Auftreten von Lokalrezidiven als Hauptproblem im Verlauf
-
ungünstigere Prognose bei Vorliegen von Gentranslokation/-fusion
7.2.2.2.2.2 Fibrosarkom [435] [436] [437]
-
Epidemiologie:
-
medianes Manifestationsalter: 50 Jahre
-
Männer bevorzugt betroffen (unabhängig von Lokalisation)
-
bei Befall der Orbita mehrere Fallberichte von Kindern
-
-
Ätiologie/Pathogenese:
-
Vorkommen als postradiogene Zweittumoren in der Orbita (z. B. nach Strahlentherapie eines Retinoblastoms)
-
erhöhtes Risiko nach Implantation von Fremdmaterial
-
-
Pathologie:
-
aggressive Tumoren (in über 80% Grad 2/3 nach FNCLCC)
-
Metastasen hauptsächlich in Lunge, Knochen und regionären Lymphknoten
-
bei Anwendung moderner Diagnostikverfahren nur einzelne Fallberichte über Fibrosarkome der Orbita
-
durch verbesserte histopathologische Diagnoseverfahren und Klassifikationssysteme für Sarkome in den letzten Jahren Fibrosarkom sehr selten gestellte Ausschlussdiagnose (< 1% aller adulten Weichteilsarkome)
-
-
Therapie:
-
Resektion mit weiten Sicherheitsabständen bis zur Exenteratio
-
Exenteratio vor allem im Falle eines Lokalrezidivs
-
Stellenwert der postoperativen Strahlen- oder Chemotherapie unklar, insbesondere bei der seltenen Lokalisation in der Orbita
-
-
Prognose:
5-Jahres-Überlebensrate ca. 55%
7.2.2.2.2.3 Retinoblastom [438] [439] [440]
-
Definition:
bösartiger Tumor in der Netzhaut des Auges, für dessen Entstehung Mutationen in beiden Allelen des Retinoblastom-Gens (lokalisiert auf Chromosom 13, Bande q14) Grundvoraussetzung sind
-
Epidemiologie:
-
Häufigkeit: ca. 1/20.000 Lebendgeburten
-
häufigstes intraokuläres Malignom bei Kindern
-
m : w+=+1 : 1
-
in der Regel Auftreten vor dem 5. Lebensjahr
-
-
Formen:
-
45% erbliche Form
-
-
heterozygot für Mutation im Retinoblastom-Gen, hinzu kommt die Mutation im zweiten Gen
-
meist beide Augen betroffen
-
durchschnittliches Alter bei Diagnosestellung: 12 Monate
-
55% nicht erbliche Form
-
-
beide Mutationen müssen in derselben Körperzelle auftreten
-
meist einseitiger Befall
-
durchschnittliches Alter bei Diagnosestellung: 23 Monate
-
75% unilateral, 25% bilateral
-
-
Klinik:
Leukokorie, Strabismus, seltener: schmerzhafte Rötung des Auges, Glaukom, Visusstörung
-
Diagnostik:
-
ophthalmologische Untersuchung: unifokale oder multifokale weißliche, vaskularisierte retinale Raumforderung, ggf. mit Tumoraussaat
-
Sonografie
-
MRT (zur Evaluierung der Beteiligung des Nervus opticus und einer eventuellen extraokulären Ausbreitung)
-
Gendiagnostik (Differenzierung zwischen hereditärer und nicht hereditärer Form)
-
in der Regel keine Biopsie
-
Lumbalpunktion bei Ausbreitung Richtung ZNS
-
ggf. Skelettszintigraphie/Knochenmarkpunktion bei Verdacht auf Knochenmetastasierung
-
-
Therapie:
-
Ziele: Lebenserhalt (primäres Ziel), Funktionserhalt (sekundäres Ziel)
-
Laser-/Kryokoagulation, transpupilläre Thermotherapie (v. a. bei kleinen Tumoren)
-
Chemotherapie als eine der wichtigsten Optionen: ermöglicht in einem beträchtlichen Teil der Fälle die Vermeidung von Strahlentherapie oder Enukleation
-
Enukleation einschließlich Resektion des Nervus opticus (v. a. bei fortgeschrittenem, unilateralem Tumor)
-
Strahlentherapie (früher Standardtherapie bei hoher Strahlensensibilität, aber signifikantes Risiko für postradiogene Zweittumoren – Risiko etwa 36% innerhalb von 50 Jahren bei hereditärer Form – daher bestimmten Indikationen vorbehalten, z. B. bei Metastasierung oder fehlendem Ansprechen auf Chemotherapie)
-
Brachytherapie: Primärtherapieoption bei solitären Tumoren ventral des Äquators (in ⅔ mit guter Sehfunktion) oder nach Versagen anderer Therapiestrategien, adjuvante Therapieoption
-
Adjuvante Chemotherapie, v. a. nach Enukleation, wenn bestimmte Risikofaktoren vorliegen (z. B. Aderhautinvasion, Sehnerveninvasion, Ausbreitung in die Orbita, Aussaat in die Vorderkammer)
-
-
Prognose:
-
5-Jahres-Überlebensrate: etwa 96% in entwickelten Ländern
-
bei hereditärer Form: erhöhtes Risiko für sekundäre (extraorbitale) Tumoren
-
7.2.2.2.2.4 Malignes Gliom des Sehnervs [412] [441] [442]
-
Definition:
entspricht dem anaplastischen Astrozytom, WHO Grad III, oder dem Glioblastom, WHO Grad IV; keine Assoziation zur Neurofibromatose
-
Epidemiologie:
-
extrem selten
-
Manifestationsgipfel: mittleres Erwachsenenalter
-
-
Symptome:
-
ähnelt zunächst einer akuten Neuritis nervi optici
-
retroorbitaler Schmerz
-
bei Diagnosestellung in 75 % bereits Beteiligung des Chiasmas, frühzeitige ZNS-Infiltration
-
rasch progredienter Visusverlust bis zur Erblindung
-
eventuell andere neurologische Symptome, z. B. Krampfanfälle
-
-
-
keine wirkungsvolle Therapie
-
kombinierte Strahlenchemotherapie mit Temzolomid
-
chirurgische Resektion nur in Einzelfällen, z. B. bei streng einseitigem Befall ohne Chiasmabeteiligung
-
-
Prognose:
Tod in der Regel innerhalb eines Jahres
Weitere Beispiele für bösartige orbitale Neoplasien nicht epithelialen Ursprungs sind in [Tab. 9] in knapper Form dargestellt.
|
Neoplasie |
Charakteristika |
|---|---|
|
▪ Epidemiologie:
▪ Pathologie:
▪ Klinik:
– Differenzialdiagnose:
|
|
|
▪ Epidemiologie:
▪ Lokalisation:
▪ Therapie:
|
|
|
▪ Epidemiologie:
▪ Pathologie:
▪ Therapie:
▪ Prognose:
|
|
▪ Epidemiologie:
|
|
|
▪ Epidemiologie:
▪ Pathologie:
▪ Therapie:
▪ Prognose:
|
|
|
Epitheloidsarkom[ 456] |
▪ Epidemiologie:
▪ Pathologie:
▪ Prognose:
|
|
Neuroblastom [423] [457] [458] [459] (bösartige Form der neuroblastischen Tumoren) |
▪ Definition:
▪ Klinik:
▪ Therapie:
|
|
maligner peripherer Nervenscheidentumor [412] [] |
|
|
orbitales Melanom [460] |
▪ Pathologie:
▪ Klinik:
▪ Diagnostik:
▪ Therapie:
|
|
endodermaler Sinuszelltumor [461] [462] (Synonym: Dottersacktumor) |
Epidemiologie:
▪ Pathologie:
▪ Klinik:
▪ Therapie:
|
7.2.2.2.3 Lymphoproliferative Erkrankungen der Orbita
7.2.2.2.3.1 Lymphome der Orbita [365] [463] [464] [465]
Anmerkung: siehe auch Kapitel 7.2.1.2.3 „Lymphome der Tränendrüse“
-
Epidemiologie:
-
Lymphome stellen die häufigsten malignen orbitalen Raumforderungen dar (etwa 50–55% aller orbitalen Malignome sind Non-Hodgkin-Lymphome)
-
Lymphome stellen bei älteren Patienten (> 60 Jahre) die häufigsten primären Orbitatumoren dar
-
lymphoide Tumoren machen etwa 10–20% der orbitalen Raumforderungen aus
-
1–2% aller Lymphome und etwa 8% aller extranodalen Lymphome betreffen die okulären Adnexe (Bindehaut, Augenlider, Tränendrüse, orbitales Weichgewebe)
-
geschätzt 5% aller Non-Hodgkin-Lymphom-Patienten haben im Verlauf der Erkrankung eine sekundäre orbitale Beteiligung
-
v. a. ältere Menschen betroffen
-
häufigste Subtypen:
-
97% der Lymphome: B-Zell-Lymphome
-
(extranodales) Marginalzonen-(B-Zell-)Lymphom (etwa 50–60%)
-
Follikuläres Lymphom (etwa 9–23%)
-
Diffus-großzelliges B-Zell-Lymphom (etwa 8–23%)
-
Mantelzell-Lymphom (etwa 5%)
-
-
Lokalisationen:
-
orbitales Weichgewebe: 32–60%
-
Konjunktiva: 15–42%
-
Tränendrüse: 14–20%
-
äußere Augenmuskeln: 9%
-
Tränensack: 2–11%
-
bilaterales Auftreten: 11–32%
-
-
Klinik:
-
Lymphome der Konjunktiva: typischerweise glatt und lachsfarben
-
bildmorphologisch manchmal schwer von der idiopathischen orbitalen Entzündung abgrenzbar (IOE, früher sog. „Pseudotumor orbitae“) → Biopsie
-
meist lokale Begrenzung okulärer Adnexlymphome bei Diagnosestellung (Ausnahme Mantelzelllymphom)
-
-
Therapie:
-
bei lokalisierter Manifestation: Strahlentherapie → gute lokale Kontrolle und gute Heilungsrate
-
bei bilateraler oder systemischer Erkrankung und aggressiven Subtypen (z. B. diffus-großzelligem B-Zell-Lymphom): Chemotherapie
-
Immuntherapie mit Anti-CD-20-Antiköpern
-
-
Prognose:
-
Low-grade-Lymphome (Marginalzonenlymphom, follikuläres Lymphom): gut
-
High-grade-Lymphome (diffus-großzelliges B-Zell-Lymphom, Mantelzelllymphom): schlecht
-
Fallbeispiel 4

-
Anamnese:
-
Erstvorstellung eines 58-jährigen, männlichen Patienten in der Augenklinik des Universitätsklinikums Halle/S.
-
Beschwerden seit zwei Jahren
-
-
Symptomatik:
-
linkes Auge vorgewölbt und gerötet
-
aktuell Visusverschlechterung
-
Den präoperativen Befund zeigt ([Abb. 20a, b])
-
bisherige Maßnahmen und Verlauf:
-
umfangreiche Diagnostik (einschließlich MRT-Untersuchungen) und Therapie durch niedergelassene Augenärztin in Zusammenarbeit mit einer großen städtischen Klinik
-
auswärtig durchgeführte transkonjunktivale Biopsie aus der Orbita: histopathologisch lymphoplasmazelluläres Infiltrat ohne auffällige IgG4-Ratio
-
Arbeitsdiagnose: idiopathische inflammatorische Orbitopathie links > rechts
-
-
therapierefraktärer Verlauf unter hochdosierten Glukokortikoiden seit etwa eineinhalb Jahren (80 mg Prednisolon zum Zeitpunkt der Erstvorstellung!), offensichtliche Cushing-Symptomatik, steroidinduzierter Diabetes mellitus, arterielle Hypertonie, Herzinsuffizienz NYHA III, Steatosis hepatis
-
Diagnostik / klinischer Befund (Auszüge):
-
kein relativer afferenter Pupillendefekt, eingeschränkte Bulbusmotilität (v. a. Adduktion) links, Doppelbilder beim Blick nach oben und zur Seite
-
vordere Augenabschnitte:
-
Bindehautgefäße / episklerale Gefäße gestaut
-
Lidkantenparallele konjunktivale Falten (LIPCOF) ++
-
-
Funduskopie:
-
links Papille temporal abgeblasst
-
Netzhaut- und Aderhautfalten
-
-
Visus:
-
rechts: 1,0
-
links: 0,4
-
Hertel-Exophthalmometrie (in mm):
23 – 123 – 28
-
Tensio:
-
rechts 15 mmHg
-
links 15 mmHg
-
-
CT ([Abb. 21a, b]):
-
links Nachweis von 4 polylobulierten, homogen mäßig KM-anreichernden raumfordernden, intra- und extrakonal gelegenen Läsionen, die größte 18,5 x 26 x 18 mm
-
rechts wesentlich kleinere intramuskuläre Läsionen
-
-

-
Verlauf:
-
zunächst Umstellung der Medikation auf eine kortisonsparende, immunmodulierende Therapie mit initial Methotrexat, bei Unverträglichkeit Umstellung auf Mycophenolatmofetil; rheumatologische Mitbetreuung
-
bei ausbleibender Besserung der Beschwerden HNO-ärztliche Mitbeurteilung und gemeinsame Indikationsstellung zur balancierten Orbitadekompression mit erneuter intraorbitaler Biopsie ([Abb. 22]) zur Überprüfung der Diagnose; Übernahme in die HNO-Abteilung
-
-
Therapie:
-
balancierte, mediale und laterale Orbitadekompression links und Biopsie einer der Raumforderungen (weitgehend intrakonal gelegen, sich nach extrakonal vorwölbend) über laterale Orbitotomie
-
Ausschleichen der medikamentösen Therapie
-
-
histopathologische Diagnose:
Extranodales Marginalzonenlymphom
-
weiterer Verlauf:
-
postoperativ leichte Besserung des Visus (0,5)
-
Staging mittels Ganzkörper-CT und cMRT ohne Anhalt für weitere Manifestationen des Lymphoms
-
Fallvorstellung im Tumorboard und Therapieübernahme durch die Kliniken für Hämatoonkologie und Strahlentherapie
-
lokale Radiotherapie bis 30,6 Gy (ED 1,8 Gy) rechte und linke Orbita
-
Anbindung an Klinik für Hämatoonkologie
-

7.2.2.2.3.2 Leukämien
-
Epidemiologie:
-
orbitale Manifestation im Vergleich zu Lymphomen deutlich seltener
-
Anteil der Leukämien an den malignen lymphoiden orbitalen Adnexerkrankungen: ca. 4% [365]
-
Auftreten meist während der ersten Lebensdekade
-
Formen:
-
orbitale Manifestation bei akuten und chronischen Leukämien möglich
-
häufigster Subtyp: akute myeloische Leukämie [466] [467] [468]
-
meist sekundäre Orbitabeteiligung (z. B. als Rezidiv), orbitale Manifestation kann aber auch Erstsymptom sein
-
kann beidseits auftreten
-
Orbitabeteiligung meist Anzeichen für fortgeschrittenes Erkrankungsstadium
-
Therapie:
Chemotherapie und Strahlentherapie des betroffenen Auges (besseres Outcome als bei alleiniger Chemotherapie) [469]
7.2.2.2.4 Metastasen [59] [470] [471]
-
Epidemiologie:
-
7–8% aller orbitalen Neoplasien
-
in bis zu 25% der Fälle vor dem Primarius evident
-
Differenzialdiagnosen:
klinisch und bildmorphologisch Verwechslung mit anderen Erkrankungen, z. B. Abszessen oder Parasitosen möglich [471]
-
Lokalisation des Primarius:
-
bei Erwachsenen vor allem in Mamma, Lunge, GI-Trakt, Prostata, Niere, Haut
-
bei Erwachsenen mit Befall der äußeren Augenmuskeln in Mamma, Haut (Melanom) [471]
-
bei Kindern v. a. Neuroblastom, Ewing-Sarkom
-
-
Therapie und Management:
-
Primärtumorsuche
-
abhängig vom Gewebetyp
-
Strahlentherapie als wesentliche therapeutische Säule (zumindest zur Symptomkontrolle)
-
-
Anamnese:
-
62-jähriger männlicher Patient
-
Seit vier Tagen wässrige Rhinorrhoe, Epiphopra, zunehmende periorbitale Schwellung und Rötung, zunehmende Schmerzen des linken Auges
-
Kein Infekt der oberen Atemwege
-
-
Befund:
-
Rötung und Schwellung des linken Ober- und Unterlids mit Ausläufern auf die Wange
-
Exophthalmus
-
Chemosis mit Bindehauthyperämie
-
Motilitätseinschränkung des Bulbus
-
Visus reduziert (0,6)
-
Augeninnendruck rechts 14, links 20 mmHg
-
Leukozyten und CRP im Blut leicht erhöht
-
CT (mit Kontrastmittel; [Abb. 23 a, b]
-
12 x 11 x 10 mm große, wandbetont kräftig KM-aufnehmende Läsion im Bereich des Musculus rectus inferior, den Nervus opticus leicht auslenkend
-
Nasennebenhöhlen regelrecht belüftet
-
-
-
Verlauf:
-
Bei Verdacht auf intraorbitalen Abszess transnasale, transethmoidale endoskopische Orbitotomie mit Eröffnung der Läsion, dabei nur wenig trübes Sekret entleert, Abstrich und Biopsie entnommen
-
Histologie: Neurodenokriner Tumor
-
Diagnose nach Primariussuche und Staging (u. a. DOTATOC-PET-CT): Neuroendokriner Tumor (G2) des Ileums mit hepatischer, orbitaler und lymphnodaler Metastasierung
-
7.2.2.2.5 Fortgeleitete (sekundäre) Orbitatumoren [59] [472]
Ursprungsort:
-
Nasennebenhöhlen
-
Einteilung: Grad I (Infiltration Orbitaknochen), Grad II (Infiltration Periorbita) und Grad III (Infiltration des orbitalen Weichgewebes durch Überschreiten der Periorbita)
-
Therapie: multimodaler Ansatz erforderlich (u. a. R0-Resektion prognoseentscheidend)
-
Prognose: bei Grad III Verbesserung der Überlebenschancen durch Exenteratio orbitae im Vergleich zum Organerhalt
-
intrakraniell
-
Augenlider (z. B. Basalzellkarzinome)
-
Bindehaut (z. B. Melanom)
7.2.3 Vaskuläre Anomalien (Gefäßanomalien)
Sie nehmen eine Sonderstellung ein und lassen sich weiter in Gefäßtumoren und Gefäßmalformationen (vaskuläre Malformationen) unterteilen.
7.2.3.1 Gefäßtumoren
Gefäßtumoren besitzen folgende Eigenschaften:
-
„echte“ Neoplasien mit endothelialer Proliferation und Angiogenese
-
Fähigkeit zur Regression (zumindest teilweise) mit zunehmendem Alter der Patienten
-
Unterscheidung in benigne, lokal aggressive (borderline) und maligne Unterformen
7.2.3.1.1 Infantiles Hämangiom [2] [473] [474] [475]
-
Synonym: Kapilläres Hämangiom (frühere Bezeichnung)
-
Epidemiologie:
-
häufigster vaskulärer Orbitatumor im Kindesalter
-
Manifestation typischerweise in den ersten Lebenswochen (nicht bei Geburt) mit Größenzunahme in den ersten 3–6 Lebensmonaten
-
Spontane Rückbildung typischerweise ab dem 6. Lebensmonat bis zum 9. Lebensjahr (im Alter von 7 Jahren in 75 – 90 % zurückgebildet [476]), selten erst in der Pubertät
-
in der Regel harmlos, bei ungünstiger Lokalisation (z. B. Orbita, Mundhöhle) aber funktionelle Beeinträchtigung und dauerhafte Deformationen möglich (z. B. irreversible Amblyopie, Astigmatismus)
-
Lokalisation und Erscheinungsbild ([Abb. 24a, b]):
-
Vorkommen als klein und umschrieben bis hin zu groß und multifokal
-
am häufigsten extrakonal gelegen, meist ventral des Bulbus im Bereich der Augenlider, extraokuläre Muskeln und Tränendrüse können mitbetroffen sein
-
seltener intrakonal gelegen, z. B. im Bereich des orbitalen Fettkörpers [477]
-
bei retrobulbärer Lage mit Gefahr der Kompression des Nervus opticus (v. a. bei schnellem Wachstum)
-
-
Klinik:

-
himbeerartiges Aussehen, v. a. bei oberflächlicher Lage
-
in der Regel harmlos, bei ungünstiger Lokalisation (z. B. Orbita, Mundhöhle) aber funktionelle Beeinträchtigung und dauerhafte Deformationen möglich (z. B. irreversible Amblyopie, Astigmatismus)
-
Differenzialdiagnose:
kongenitales Hämangiom
-
viel seltener, in der Orbita absolute Rarität
-
bereits bei Geburt vorhanden und vollständig ausgebildet (kein Wachstum nach der Geburt)
-
Therapie [478]:
-
spontane Remission in 85–90% der Fälle
-
Therapie nur bei symptomatischen Hämangiomen notwendig; bei Kompression des Nervus opticus rasche Therapie erforderlich
-
Propranolol 2–3 mg / kg KG / Tag verteilt auf 2–4 Einzeldosen für 6 Monate (Therapie der ersten Wahl bei komplizierten infantilen Hämangiomen, Ansprechen: 98 %)
-
systemische Steroidgabe inzwischen Ausnahmefällen vorbehalten (z. B. bei Atemwegsverlegung, Nichtansprechen anderer Therapieverfahren)
-
Zytostatika (Vincristin) und Interferon-alpha obsolet
-
Laser-/Kryotherapie: bei kleinen, planen, gut zugänglichen Hämangiomen (z. B. Lid)
-
chirurgische Resektion: im Bereich der Orbita eher selten indiziert, da Gefahr der Schädigung umgebender Strukturen
-
7.2.3.1.2 Hämangioperizytom [479] [480]
-
Definition:
von den Perizyten der Gefäßwand oder pluripotenten perivaskulären Zellen ausgehender Tumor
-
Epidemiologie:
nur Einzelfallberichte zu Vorkommen in der Orbita [480]
-
Diagnostik: [480]
-
keine pathognomonischen klinischen oder radiologischen Merkmale
-
Biopsie erforderlich
-
-
Pathologie:
-
histologisch gesamte Bandbreite benigner bis maligner Eigenschaften → Einteilung in benigne, borderline und maligne
-
Verhalten eher unberechenbar, auch scheinbar gutartige Läsionen zeigen gelegentlich malignes Verhalten
-
Metastasierung in ca. 15% der Fälle
-
Hämangioperizytome und solitäre fibröse Tumoren haben viele histologische und immunhistochemische Überschneidungen und stellen laut einiger Autoren möglicherweise eine gemeinsame Entität dar [479]
-
-
Therapie:
-
vollständige chirurgische Resektion
-
ggf. adjuvante Strahlentherapie
-
ggf. Chemotherapie
-
Follow-up
-
-
Prognose:
Lokalrezidive in ca. 30% der Fälle
In [Tab. 10] sind Beispiele für weitere orbitale Gefäßtumoren aufgeführt. Die meisten davon sind absolute Raritäten in der Orbita und es gibt oft nur einzelne Fallberichte [475]. Das Büschel-Angiom wird inzwischen von vielen Experten zu den Borderline-Tumoren gezählt und bildet mit dem kaposiformen Hämangioendotheliom eine gemeinsame Entität – mit einem histopathologischen Spektrum, das, je nach Einzelfall, mehr oder weniger aggressive Merkmale aufweisen kann [494] [495].
|
Dignität |
Gefäßtumoren |
|---|---|
|
gutartig |
|
|
lokal aggressiv / borderline |
|
|
bösartig |
|
*Das Büschel-Angiom erscheint in der ISSVA-Klassifikation bei den gutartigen Tumoren, ist aber als Borderline-Tumor zu erachten, der mit dem kaposiformen Hämangioendotheliom eine gemeinsame Entität bildet.
7.2.3.2 Gefäßmalformationen [2] [473] [474] [475] [495]
Vaskuläre Malformationen besitzen folgende Eigenschaften:
-
mesenchymale, vaskulogenetische Entwicklungsstörungen mit geringer proliferativer Aktivität
-
stets bei Geburt vorhanden, aber oft erst später auffallend (Wachstum z. B. hormonell oder mechanisch getriggert)
-
Progress
-
keine Spontanremission
Eine Einteilung der Gefäßmalformationen und Therapieoptionen sind in [Tab. 11] dargestellt. Die prozentualen Häufigkeitsangaben sind allgemein und nicht orbitaspezifisch [2] [475].([Abb. 25a, b])

|
Einteilung |
Therapie [474] |
|---|---|
|
Einfache vaskuläre Malformationen |
|
|
Slow-flow |
|
|
▪ venöse Malformation (70 %) |
Sklerotherapie, Lasertherapie, Resektion, ggf. mTOR-Inhibitoren |
|
▪ lymphatische Malformation (12 %)
|
Sklerotherapie (makrozystische Form), Lasertherapie, Resektion, mTOR-Inhibitoren (Sirolimus) [488] |
|
▪ Kapilläre Malformation |
Lasertherapie |
|
Fast-flow |
|
|
▪ arteriovenöse Malformation (8 %) |
Embolisation (transarteriell, transvenös, direkt), Resektion des okkludierten Nidus (= Kern, der die Shuntgefäße enthält) |
|
▪ arteriovenöse Fistel |
Embolisation |
|
▪ kombinierte vaskuläre Malformationen (6%) |
|
|
7.2.3.2.1 Venöse Malformation [201] [412]
-
Synonym:
früher „kavernöses Hämangiom“, laut aktueller ISSVA-Klassifikation vaskulärer Anomalien [494] nur noch als „slow-flow venöse Malformationen“ zu bezeichnen (alte Bezeichnung allerdings weiterhin sehr verbreitet)
-
Epidemiologie:
-
einer der häufigsten benignen Raumforderungen der Orbita beim Erwachsenen (5–7% aller orbitalen Raumforderungen)
-
typisches Manifestationsalter: 30–50 Jahre
-
Frauen häufiger betroffen
-
-
Klinik:
-
nicht selten Zufallsbefund
-
langsames Wachstum (sich über Jahre entwickelnder Exophthalmus)
-
verstärktes Wachstum während der Schwangerschaft
-
typische Lokalisation: intrakonal
-
-
Therapie:
-
„watch and wait“ bei kleinen/asymptomatischen Befunden
-
Sklerotherapie
-
komplette chirurgische Resektion, z. B. über laterale Orbitotomie
-
-
Differenzialdiagnose:
„Orbitale Varizen“
-
Begriff „Orbitale Varizen“ inzwischen veraltet, wird den venösen Malformationen zugeordnet
-
Pathogenese: angeborene Schwäche der Venenwände der klappenlosen Orbitalvene → massive Erweiterung
-
Diagnostik: Exophthalmus während Valsalva-Manöver
-
Klinik: bei Einblutung schnelle, schmerzhafte Zunahme des Exophthalmus
-
Therapie: in der Regel keine erforderlich
7.2.3.2.2 Lymphatische Malformation [412] [473]
-
Synonym (veraltet):
Lymphangiom
-
Epidemiologie:
v. a. im Kindes- und Jugendalter auftretend
-
Klinik:
-
periorbitale Schwellung
-
langsam progredienter, schmerzloser Exophthalmus
-
Motilitätsstörungen
-
Bulbusverlagerung
-
Komplikation: akute Exazerbation durch Einblutung oder Infektion möglich (starke Vergrößerung innerhalb von Stunden oder Tagen möglich)
-
keine Kapsel, diffus infiltrierend, Kompartimentgrenzen nicht respektierend
-
mit Lymphe oder auch Blut gefüllt
-
-
Therapie:
-
vollständige Exstirpation i. d. R. nicht möglich
-
„watch and wait“
-
bei drohendem Visusverlust volumenreduzierende Maßnahmen, z. B. Teilresektion, Injektion sklerosierender Substanzen, mTOR-Inhibitoren
-
7.2.3.2.3 Arteriovenöse Malformation (AVM) [473] [475] [495] [496]
-
Lokalisation:
-
v. a. Kopf-Hals-Bereich, inklusive Gehirn ([Abb. 26])
-
Extremitäten
-
seltener innere Organe (Leber, Lunge …)
-

-
Eigenschaften:
-
klinisch sehr vielgestaltig und herausfordernd
-
ausnahmslos lebenslanges progressives Wachstum (destruktiv!)
-
reagieren auf Stimuli wie hormonelle Veränderungen, (chirurgische) Traumata (→ vermehrte Proliferation nach unvollständiger invasiver Therapie möglich!)[474]
-
-
klinische Einteilung (Schobinger-Klassifikation, verkürzt) [497]:
-
Stadium I: klinisch inaktiv
-
Stadium II: Expansion, Pulsation
-
Stadium III: destruktive AVM mit Ulzerationen, Nekrose, Blutung, Schmerz, Infektion
-
Stadium IV: dekompensierte AVM mit Herzinsuffizienz
-
-
Therapie:
-
oft sehr schwierig und risikobehaftet
-
„watch and wait“ bei asymptomatischen AVM
-
möglichst vollständiger Verschluss durch Embolisation (mehrere Sitzungen) mit anschließender
-
Resektion (sofern angesichts der Lokalisation möglich), um Rezidive zu verhindern
-
mTOR-Inhibitoren nicht wirksam
-
-
Prognose:
hohe Rezidivneigung bei inkompletter Therapie
Fallbeispiel 6



-
Anamnese:
-
notfallmäßige Vorstellung eines 9 Monate alten Jungen mit akutem Exophthalmus rechts
-
keine relevanten Vorerkrankungen
-
-
Diagnostik:
-
Inspektion: Exophthalmus rechts, vermehrte Gefäßinjektion, Lidschluss unvollständig ([Abb. 27])
-
Sonografie bei Erstvorstellung: Verdacht auf akut eingeblutete, kavernöse venöse Malformation retrobulbär, MRT zur weiteren Abklärung empfohlen
-
MRT ([Abb. 28]): retrobulbäre Einblutung bei V. a. rupturiertes, teilthrombosiertes Aneurysma spurium mit zusätzlichen venösen Malformationsanteilen, DD AV-Malformation
-
Angiografie: High-flow arteriovenöse Malformation in der Orbita rechts, hauptsächlich gespeist aus der Arteria ophthalmica rechts (…) dominanter venöser Ausstrom über dilatierte venöse Aneurysmen (Typ II nach WHO-Klassifikation). Venöse Drainage über Sinus cavernosus.
-
-
Verlauf:
-
progredienter Exophthalmus rechts
-
zunehmende Abwehrhaltung des Kindes
-
ausgeprägte Visusminderung rechts
-
-
Therapie:
-
Embolisation der arteriovenösen Malformation mit Onyx18 (Interventionelle Radiologie) → vollständiger Verschluss des Zuflusses über die A. ophthalmica und eines weiteren Feeders aus der A. maxillaris → AVM somit operationsfähig.
-
bei progredientem Exophthalmus und nun Hornhautulzeration des rechten Auges interdisziplinäre Fallbesprechung und Indikation zur operativen Therapie
-
Enucleatio bulbi, Resektion der intrakonalen AVM nach Embolisation, Implantation einer Guthoffplombe (16 mm) mit Tenon- und Bindehautplastik (interdisziplinär durch Augenheilkunde und HNO; [Abb. 29])
-
spätere epithetische Versorgung
-
-
Histologie:
-
arteriovenöse Malformation bei Z. n. Embolisation
-
kein Anhalt für Malignität
-

7.3. Konklusion
Neoplasien der Orbita sind selten, aber sehr vielfältig und anhand der Symptomatik allein kaum von strukturellen oder entzündlichen Läsionen zu unterschieden. Hierfür ist eine differenzierte ophthalmologische, radiologische und paraklinische Diagnostik wichtig – wobei jedoch meist das histopathologische Untersuchungsergebnis der Probebiopsie entscheidend ist. Eine Ausnahme besteht dabei für Tumoren epithelialen Ursprungs der Tränendrüse, bei denen sich durch Inzisionsbiopsien die Gesamtprognose signifikant verschlechtert. Die Einteilung von Tumoren der Orbita erfolgt in Neoplasien epithelialen und nicht epithelialen Ursprungs sowie Neoplasien des lymphatischen Gewebes, wobei die Tränendrüse das einzige Organ der Orbita mit epithelialen Strukturen darstellt. Eine Sonderstellung haben vaskuläre Malformationen, welche ein sehr variables klinisches Verhalten aufweisen. Therapeutisch kommt neben der Watch-and-wait-Strategie bei asymptomatischen, benignen Prozessen vor allem die chirurgische Resektion bei symptomatischen, benignen Neoplasien in Betracht, wobei gerade im Bereich der Orbita ganz besonders zwischen dem zu erwartenden Benefit und möglichen funktionellen Defiziten abgewogen werden muss. Die Zugangswege sind dabei vor allem von der Lokalisation der Raumforderung abhängig. Die Strahlentherapie sowie medikamentöse Systemtherapien mit Chemotherapeutika und Biologika sind wesentliche therapeutische Säulen und haben in den letzten Jahrzehnten die Morbidität von Patienten mit orbitalen Tumoren reduzieren können. Trotzdem bleibt festzuhalten, dass bösartige Tumoren der Orbita zumeist mit einer schlechten Gesamtprognose einhergehen.
Lymphoproliferative Läsionen der Orbita sind indolent, so dass Patienen bei Remission häufig eine gute Lebensqualität haben, trotzdem sollte man sich über die z. T. immer noch hohen Mortalitätsraten, z. B. bei NK-/T-Zell-Lymphomen, bewusst sein. Ein besseres Verständnis der molekulargenetischen Zusammenhänge wird zukünftig zu einem präziseren Einsatz von Targettherapeutika und damit zu einer besseren Prognose von Lymphomen der Orbita führen. Grundvorraussetzung hierfür sind allerdings Biopsien, die wirklich repräsenativ sind.
Knöcherne Tumoren der Orbita beinhalten sehr unterschiedliche Entitäten, wobei die fibro-ossären Läsionen faszinierende pathogenetische und pathologische Eigenschaften haben. Duch die Bildgebung lässt sich die Liste der Differenzialdiagnosen eingrenzen, zur Diagnosestellung ist aber eine Biopsie oft notwendig, deren Beurteilung meist Referenzzentren vorbehalten bleibt. Man sollte sich bei der Therapieentscheidung sowohl über systemische Auswirkungen der Erkrankung als auch über neue Ansätze der Targettherapien bei diesen Tumoren bewusst sein.
Tumoren der Tränendrüse bleiben aufgrund ihrer Seltenheit und Diversität nicht nur für den Pathologen, sondern auch für den Kliniker eine echte Herausforderung. Auch wenn die Anamnese und das Erfassen der Symptome wichtig für die Diagnosestellung sind, können diese doch stark variieren und auch die Bildgebung ist selten pathognomonisch. Weiterhin gibt es Kontroversen über die geeigneten Therapieansätze, insbesondere bei malignen epithelialen Tumoren, welche generell im Vergleich zu Malignomen der Speicheldrüsen eine schlechtere Prognose haben.
Primäre und sekundäre neurogene Tumoren sind häufig in der Orbita lokalisiert, vermutlich durch die hohe Dichte an Nerven, welche in dieser Region verlaufen. Diagnostik und Therapie stellen eine Herausforderung dar, da weiterhin Unklarheiten bezüglich der Ätiologie, Pathogenese und dem natürlichen Krankheitsverlauf bestehen.
Tumoren des orbitalen Fett- und Bindegewebes sowie der quergesteiften Muskulatur sind zumeist seltene heterogene Neoplasien, welche jedoch in der klinischen Symptomtik, histogenetisch, molekularbiologisch sowie prognostisch Ähnlichkeiten untereinander aufweisen.
Metastasen der Orbita sind eine ebenso heterogene Gruppe von Neoplasien, deren Präsentation je nach Ausdehnung der Erkrankung und Biologie des Primärtumors variiert. Die meisten Metastasen kommen bei Patienten mit bekanntem Primärtumor vor und machen sich durch Abweichung des Bulbus, Strabismus und Schmerzen bemerkbar. Bei Verdacht sind Bildgebung und Biopsie zur Planung einer multidisziplinären Behandlung notwendig, mit dem Ziel, den Visus zu erhalten, Schmerzen zu reduzieren und die Lebensqualität zu verbessern. In einigen Fällen ist hierbei ein kurativer Ansatz möglich.
Vaskuläre Anomalien werden in Gefäßtumoren und vaskuläre Malformationen unterteilt. Während es sich bei den Erstgenannten um „echte“ Neoplasien mit endothelialer Proliferation und Angiogenese handelt, sind Malformationen vaskulogenetische Entwicklungsstörungen. Diese zeigen eine geringe proliferative Aktivität, sind zwar bereits bei Geburt vorhanden, werden aber meist erst später apparent und zeigen keine Spontanremission. Die heterogene klinische Präsentation von Gefäßtumoren erfordert die Abklärung verschiedener Differenzialdiagnosen. Es existiert ein großes Spektrum an Therapiealternativen wie medikamentösen, Laser- und klassisch-chirurgischen Interventionen, sodass die meisten Patienten von einer der Optionen profitieren. Durale AV-Fisteln und Carotis-cavernosus-Fisteln können variable orbitale Manifestationen haben. Trotz der Unterschiedlichkeit der Läsionen kommen die gleichen Untersuchungsmethoden zum Einsatz. Charakterisierung der Läsion und Therapieplanung erfordern eine konventionelle Katheterangiographie. Bei benignen Fällen ist die Therapiestrategie konservativ, bei rasch progredienten und schweren Fällen ist ein neuroendovaskuläres Management zu favorisieren.
Interessenkonflikt
Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
8. Danksagung
Die Autoren möchten sich ganz herzlich bei Herrn Prof. H.-J. Welkoborsky, Frau Dr. C. Adderson-Kisser, Herrn Dr. I. Seiwerth, Frau A. Gey, Herrn Prof. W. Wohlgemuth, Frau A. Troch und Herrn Prof. S. Plontke für ihre Unterstützung bei der Erstellung des Manuskripts bedanken.
-
Literatur
- 1 Poloschek CM, Lagreze WA, Ridder GJ. et al. [Clinical and neuroradiological diagnostics of orbital tumors]. Ophthalmologe 2011; 108: 510-518
- 2 Plontke SK, Glien A, Kisser U. et al. [Diseases and Surgery of the Orbit]. Laryngorhinootologie 2020; 99: 896-917
- 3 AWMF. Rekonstruktion von Orbitadefekten (S2e-Leitlinie), Registernummer: 007/099. In: Arbeitsgemeinschaft medizinisch wissenschaftlicher Fachgesellschaften (AWMF); 2013
- 4 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 57–77
- 5 Bonavolonta G, Tranfa F, de Conciliis C. et al. Dermoid cysts: 16-year survey. Ophthalmic Plast Reconstr Surg 1995; 11: 187-192
- 6 Jordan DR, Spitellie P, Brownstein S. et al. Orbital cholesterol granuloma and cholesteatoma: significance of differentiating the two. Ophthalmic Plast Reconstr Surg 2007; 23: 415-417
- 7 Sherman RP, Rootman J, Lapointe JS. Orbital dermoids: clinical presentation and management. Br J Ophthalmol 1984; 68: 642-652
- 8 Shields JA, Kaden IH, Eagle RC. et al. Orbital dermoid cysts: clinicopathologic correlations, classification, and management. The 1997 Josephine E. Schueler Lecture. Ophthalmic Plast Reconstr Surg 1997; 13: 265-276
- 9 Delfini R, Missori P, Iannetti G. et al. Mucoceles of the paranasal sinuses with intracranial and intraorbital extension: report of 28 cases. Neurosurgery 1993; 32: 901-906 discussion 906
- 10 Heichel J, Struck HG, Hammer T. et al. [Pediatric acute dacryocystitis due to frontoethmoidal mucocele]. HNO 2019; 67: 458-462
- 11 Nicollas R, Facon F, Sudre-Levillain I. et al. Pediatric paranasal sinus mucoceles: etiologic factors, management and outcome. Int J Pediatr Otorhinolaryngol 2006; 70: 905-908
- 12 Holtmann C, Brachert M, Spaniol K. et al. [Dacryolith in a lacrimal duct]. Ophthalmologe 2016; 113: 244-247
- 13 Tanaboonyawat S, Idowu OO, Copperman TS. et al. Dacryops - A review. Orbit 2020; 39: 128-134
- 14 Junemann A, Holbach LM. [Epithelial giant inclusion cyst 50 years after enucleation without orbital implant]. Klin Monbl Augenheilkd 1998; 212: 127-128
- 15 Kalantzis GK, Verity DH, Rose GE. Periocular implantation cysts: a late complication of ophthalmic surgery. Eye (Lond) 2014; 28: 1004-1007
- 16 Gupta R, Seith A, Guglani B. et al. Congenital cystic eye: features on MRI. Br J Radiol 2007; 80: e137-140
- 17 Guthoff R, Klein R, Lieb WE. Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 2004; 242: 268-271
- 18 Kuchle HJ, Normann J, Lubbering I. [Congenital cystic eye]. Klin Monbl Augenheilkd 1986; 188: 239-241
- 19 Schittkowski MP, Guthoff RF. Injectable self inflating hydrogel pellet expanders for the treatment of orbital volume deficiency in congenital microphthalmos: preliminary results with a new therapeutic approach. Br J Ophthalmol 2006; 90: 1173-1177
- 20 Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol 2010; 94: 487-493
- 21 Badtke G, Tost M. Anophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 328–344
- 22 Badtke G, Tost M. Mikrophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 312–328
- 23 Chaudhry IA, Arat YO, Shamsi FA. et al. Congenital microphthalmos with orbital cysts: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2004; 20: 452-457
- 24 Oguz H, Ozturk A, San I. Congenital nasolacrimal duct occlusion with clinical anophthalmos: a possible new association. Ophthalmic Genet 2003; 24: 181-185
- 25 Pejic M, Luecke K, Meoded A. et al. Pediatric Cephaloceles: A Multimodality Review. Appl Radiol 2020; 49: 26-32
- 26 Terry A, Patrinely JR, Anderson RL. et al. Orbital meningoencephalocele manifesting as a conjunctival mass. Am J Ophthalmol 1993; 115: 46-49
- 27 Vashisht S, Ghai S, Hatimota P. et al. Cystic lesions of the orbit : A CT spectrum. Indian Journal of Radiology and Imaging 2003; 13: 139
- 28 Modi P, Shah NA, Bhalodia JN. et al. ORBITAL TUMORS IN CHILDREN: A DESCRIPTIVE STUDY AT TERTIARY CARE CENTRE. The Journal of medical research 2013; 3: 362-366
- 29 Green WR, Zimmerman LE. Ectopic lacrimal gland tissue. Report of eight cases with orbital involvement. Arch Ophthalmol 1967; 78: 318-327
- 30 Scheiner AJ, Frayer WC, Rorke LB et al. Ectopic brain tissue in the orbit. Eye (Lond) 1999; 13: ( Pt 2) 251–254
- 31 Damato PJ, Damato FJ. Neonatal Orbital Teratoma. Br J Ophthalmol 1962; 46: 685-691
- 32 Herman TE, Vachharajani A, Siegel MJ. Massive congenital orbital teratoma. J Perinatol 2009; 29: 396-397
- 33 Sesenna E, Ferri A, Thai E. et al. Huge orbital teratoma with intracranial extension: a case report. J Pediatr Surg 2010; 45: e27-31
- 34 Duke-Elder S. Anomalies of the Bony Orbit. In: Duke-Elder S, Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 942-949
- 35 Exner S, Bogusch G, Sokiranski R. Cribra orbitalia visualized in computed tomography. Ann Anat 2004; 186: 169-172
- 36 Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol 2004; 88: 833-834
- 37 Duke-Elder S. Deformities of the Face.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964; 1007–1035
- 38 Kawamoto HK. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg 1976; 3: 529-572
- 39 Fearon JA. Rare craniofacial clefts: a surgical classification. J Craniofac Surg 2008; 19: 110-112
- 40 Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg 1976; 4: 69-92
- 41 Heike CL, Hing AV, Aspinall CA. et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet 2013; 163C: 271-282
- 42 Dupont S, Catala M, Hasboun D. et al. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology 1997; 48: 1013-1018
- 43 Fea AM, Aragno V, Briamonte C. et al. Parry Romberg syndrome with a wide range of ocular manifestations: a case report. BMC Ophthalmol 2015; 15: 119
- 44 Duke-Elder S. Deformities of the Skull.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 1035–1072
- 45 Freudlsperger C, Castrillón-Oberndorfer G, Hoffmann J. et al. Isolierte, nichtsyndromale Kraniosynostosen. Der MKG-Chirurg 2013; 6: 301-313
- 46 Governale LS. Craniosynostosis. Pediatr Neurol 2015; 53: 394-401
- 47 Kotrikova B, Muhling J. [Surgery of craniofacial deformities]. Klin Monbl Augenheilkd 2004; 221: 970-977
- 48 Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr 2017; 29: 622-628
- 49 Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999; 36: 177-182
- 50 Dubourg C, Bendavid C, Pasquier L. et al. Holoprosencephaly. Orphanet J Rare Dis 2007; 2: 8
- 51 Lustig LR, Holliday MJ, McCarthy EF. et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg 2001; 127: 1239-1247
- 52 Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5
- 53 Lannon DA, Earley MJ. Cherubism and its charlatans. Br J Plast Surg 2001; 54: 708-711
- 54 Grass S, Welkoborsky HJ, Mobius H. et al. [Orbital complications]. HNO 2018; 66: 800-811
- 55 Lehnerdt G, Peraud A, Berghaus A. et al. [Orbital and intracranial complications of acute sinusitis. Diagnostics and therapy in children and adolescents]. HNO 2011; 59: 75-86 quiz 87-78
- 56 Stammberger H. [Complications of inflammatory paranasal sinus diseases including iatrogenically-induced complications]. Eur Arch Otorhinolaryngol Suppl 1993; 1: 61-102
- 57 Welkoborsky HJ, Grass S, Deichmuller C. et al. Orbital complications in children: differential diagnosis of a challenging disease. Eur Arch Otorhinolaryngol 2015; 272: 1157-1163
- 58 Hamed-Azzam S, AlHashash I, Briscoe D. et al. Rare Orbital Infections ~ State of the Art ~ Part II. J Ophthalmic Vis Res 2018; 13: 183-190
- 59 Rootman J. Diseases of the orbit.Second edition Aufl. Lippincott Williams & Wilkins; Philadelphia: 2003
- 60 Madge SN, Prabhakaran VC, Shome D. et al. Orbital tuberculosis: a review of the literature. Orbit 2008; 27: 267-277
- 61 Fay A, Dolman PJ. Diseases and Disorders of the Orbit and Ocular Adnexa Expert Consult. In Fay A, Dolman PJ (eds). Elsevier; 2017
- 62 Heichel J, Griebsch M, Siebolts U. et al. [Bilateral periocular necrotizing fasciitis with unilateral orbital involvement]. Ophthalmologe 2020; 117: 1121-1125
- 63 Huber L, Budjan J, Rotter N. et al. [Necrotizing fasciitis in the head and neck region-three case reports and a review of the literature]. HNO 2020; 68: 935-943
- 64 Amrith S, Hosdurga Pai V, Ling WW. Periorbital necrotizing fasciitis -- a review. Acta Ophthalmol 2013; 91: 596-603
- 65 Wong CH, Chang HC, Pasupathy S. et al. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85: 1454-1460
- 66 Lazzeri D, Lazzeri S, Figus M. et al. Periorbital necrotising fasciitis. Br J Ophthalmol 2010; 94: 1577-1585
- 67 Wladis EJ, Levin F, Shinder R. Clinical Parameters and Outcomes in Periorbital Necrotizing Fasciitis. Ophthalmic Plast Reconstr Surg 2015; 31: 467-469
- 68 Federspil P. German Society of Oto-Rhino-Laryngology H, Neck S. [Guidelines: Antibiotic treatment of infections of the head and neck: consensus report on behalf of the presidency of the German Society of Oto-Rhino-Laryngology, Head and Neck Surgery edited by P. Federspil, Homburg/Saar]. . HNO 2009; 57: 377-394
- 69 Barrani M, Massei F, Scaglione M. et al. Unusual onset of a case of chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol Online J 2015; 13: 60
- 70 Dagan O, Barak Y, Metzker A. Pyoderma gangrenosum and sterile multifocal osteomyelitis preceding the appearance of Takayasu arteritis. Pediatr Dermatol 1995; 12: 39-42
- 71 Huber AM, Lam PY, Duffy CM. et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr 2002; 141: 198-203
- 72 Hu AC, Ng WKY. Orbital Osteomyelitis in the Pediatric Patient. J Craniofac Surg 2021; 32: 206-209
- 73 Cornely OA, Alastruey-Izquierdo A, Arenz D. et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect Dis 2019; 19: e405-e421
- 74 Bakhshaee M, Bojdi A, Allahyari A. et al. Acute invasive fungal rhinosinusitis: our experience with 18 cases. Eur Arch Otorhinolaryngol 2016; 273: 4281-4287
- 75 van Burik JA, Hare RS, Solomon HF. et al. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42: e61-65
- 76 Songu M, Unlu HH, Gunhan K. et al. Orbital exenteration: A dilemma in mucormycosis presented with orbital apex syndrome. Am J Rhinol 2008; 22: 98-103
- 77 Nicolai P, Lombardi D, Tomenzoli D. et al. Fungus ball of the paranasal sinuses: experience in 160 patients treated with endoscopic surgery. Laryngoscope 2009; 119: 2275-2279
- 78 Johnson TE, Casiano RR, Kronish JW. et al. Sino-orbital aspergillosis in acquired immunodeficiency syndrome. Arch Ophthalmol 1999; 117: 57-64
- 79 Levin LA, Avery R, Shore JW. et al. The spectrum of orbital aspergillosis: a clinicopathological review. Surv Ophthalmol 1996; 41: 142-154
- 80 Siddiqui AA, Shah AA, Bashir SH. Craniocerebral aspergillosis of sinonasal origin in immunocompetent patients: clinical spectrum and outcome in 25 cases. Neurosurgery 2004; 55: 602-611 discussion 611-603
- 81 Nielsen EW, Weisman RA, Savino PJ. et al. Aspergillosis of the sphenoid sinus presenting as orbital pseudotumor. Otolaryngol Head Neck Surg 1983; 91: 699-703
- 82 Sivak-Callcott JA, Livesley N, Nugent RA. et al. Localised invasive sino-orbital aspergillosis: characteristic features. Br J Ophthalmol 2004; 88: 681-687
- 83 Lamoth F, Calandra T. Early diagnosis of invasive mould infections and disease. J Antimicrob Chemother 2017; 72: i19-i28
- 84 McCarthy M, Rosengart A, Schuetz AN. et al. Mold infections of the central nervous system. N Engl J Med 2014; 371: 150-160
- 85 Green WR, Font RL, Zimmerman LE. Asperillosis of the orbit. Report of ten cases and review of the literature. Arch Ophthalmol 1969; 82: 302-313
- 86 Denning DW. Therapeutic outcome in invasive aspergillosis. Clin Infect Dis 1996; 23: 608-615
- 87 Robert Koch-Institut. Infektionsepidemiologisches Jahrbuch meldepflichtiger Krankheiten für 2019 Berlin: Robert Koch-Institut; 2020
- 88 Gomez Morales A, Croxatto JO, Crovetto L. et al. Hydatid cysts of the orbit. A review of 35 cases. Ophthalmology 1988; 95: 1027-1032
- 89 Xiao A, Xueyi C. Hydatid cysts of the orbit in Xinjiang: a review of 18 cases. Orbit 1999; 18: 151-155
- 90 Kars Z, Kansu T, Ozcan OE. et al. Orbital echinococcosis. Report of two cases studied by computerized tomography. J Clin Neuroophthalmol 1982; 2: 197-199
- 91 Chtira K, Benantar L, Aitlhaj H. et al. The surgery of intra-orbital hydatid cyst: a case report and literature review. Pan Afr Med J 2019; 33: 167
- 92 Senyuz OF, Yesildag E, Celayir S. Albendazole therapy in the treatment of hydatid liver disease. Surg Today 2001; 31: 487-491
- 93 Pushker N, Bajaj MS, Balasubramanya R. Disseminated cysticercosis involving orbit, brain and subcutaneous tissue. J Infect 2005; 51: e245-248
- 94 Walrath JD, Lalin SC, Leib ML. Cysticercosis isolated to the orbit. Ophthalmic Plast Reconstr Surg 2003; 19: 243-244
- 95 Sotelo J, Escobedo F, Rodriguez-Carbajal J. et al. Therapy of parenchymal brain cysticercosis with praziquantel. N Engl J Med 1984; 310: 1001-1007
- 96 Nentwich MM, Pleyer U, Schaller UC. et al. [International ophthalmology and travel medicine]. Ophthalmologe 2016; 113: 83-94
- 97 Mombaerts I, Rose GE, Garrity JA. Orbital inflammation: Biopsy first. Surv Ophthalmol 2016; 61: 664-669
- 98 Ben Simon GJ, Yoon MK, Atul J. et al. Clinical manifestations of orbital mass lesions at the Jules Stein Eye Institute, 1999-2003. Ophthalmic Surg Lasers Imaging 2006; 37: 25-32
- 99 Nölle B, Both M. Idiopathische Orbitaentzündung.In Pleyer U , Hrsg. Entzündliche Augenerkrankungen. Berlin, Heidelberg: Springer- Verlag; 2016: 48–57
- 100 Swamy BN, McCluskey P, Nemet A. et al. Idiopathic orbital inflammatory syndrome: clinical features and treatment outcomes. Br J Ophthalmol 2007; 91: 1667-1670
- 101 Fujii H, Fujisada H, Kondo T. et al. Orbital pseudotumor: histopathological classification and treatment. Ophthalmologica 1985; 190: 230-242
- 102 Mombaerts I, Goldschmeding R, Schlingemann RO. et al. What is orbital pseudotumor?. Surv Ophthalmol 1996; 41: 66-78
- 103 Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982; 89: 1040-1048
- 104 Rootman J, McCarthy M, White V. et al. Idiopathic sclerosing inflammation of the orbit. A distinct clinicopathologic entity. Ophthalmology 1994; 101: 570-584
- 105 Iaconetta G, Stella L, Esposito M. et al. Tolosa-Hunt syndrome extending in the cerebello-pontine angle. Cephalalgia 2005; 25: 746-750
- 106 Speth MM, Metternich FU, Mina R. et al. Tolosa-Hunt-Syndrom, das sich als migrierende, auf Kortikosteroid reagierende CN3- und CN6-Lähmung bei verschatteten Keilbeinhöhlen darstellt. forum hno: omnimed Verlag 05/2020;
- 107 Halabi T, Sawaya R. Successful Treatment of Tolosa-Hunt Syndrome after a Single Infusion of Infliximab. J Clin Neurol 2018; 14: 126-127
- 108 Mormont E, Laloux P, Vauthier J. et al. Radiotherapy in a case of Tolosa-Hunt syndrome. Cephalalgia 2000; 20: 931-933
- 109 Perez CA, Evangelista M. Evaluation and Management of Tolosa-Hunt Syndrome in Children: A Clinical Update. Pediatr Neurol 2016; 62: 18-26
- 110 Zhang X, Zhang W, Liu R. et al. Factors that influence Tolosa-Hunt syndrome and the short-term response to steroid pulse treatment. J Neurol Sci 2014; 341: 13-16
- 111 Espinoza GM, Liu JL. Orbital Vasculitides-Differential Diagnosis. Curr Rheumatol Rep 2019; 21: 54
- 112 Tiegs-Heiden CA, Eckel LJ, Hunt CH. et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014; 35: 1393-1397
- 113 Erdei A, Steiber Z, Molnar C. et al. Exophthalmos in a young woman with no graves' disease - a case report of IgG4-related orbitopathy. BMC Ophthalmol 2018; 18: 5
- 114 Jurkov M, Olze H, Klauschen F. et al. [IgG4-Related Orbitopathy as an Important Differential Diagnosis of Advanced Silent Sinus Syndrome. German version]. HNO 2020; 68: 864-868
- 115 Mulholland GB, Jeffery CC, Satija P. et al. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44: 24
- 116 Detiger SE, Karim AF, Verdijk RM. et al. The treatment outcomes in IgG4-related orbital disease: a systematic review of the literature. Acta Ophthalmol 2019; 97: 451-459
- 117 Plaza JA, Garrity JA, Dogan A. et al. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol 2011; 129: 421-428
- 118 Perumal B, Black EH, Levin F. et al. Non-infectious orbital vasculitides. Eye (Lond) 2012; 26: 630-639
- 119 Sfiniadaki E, Tsiara I, Theodossiadis P. et al. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther 2019; 8: 227-234
- 120 Mukhtyar C, Flossmann O, Hellmich B. et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67: 1004-1010
- 121 Holle JU, Voigt C, Both M. et al. Orbital masses in granulomatosis with polyangiitis are associated with a refractory course and a high burden of local damage. Rheumatology (Oxford) 2013; 52: 875-882
- 122 Perry SR, Rootman J, White VA. The clinical and pathologic constellation of Wegener granulomatosis of the orbit. Ophthalmology 1997; 104: 683-694
- 123 Chang SY, Keogh KA, Lewis JE. et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013; 44: 2432-2437
- 124 Martinez Del Pero M, Chaudhry A, Jones RB. et al. B-cell depletion with rituximab for refractory head and neck Wegener's granulomatosis: a cohort study. Clin Otolaryngol 2009; 34: 328-335
- 125 Takanashi T, Uchida S, Arita M. et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophthalmology 2001; 108: 1129-1133
- 126 Cottin V, Cordier JF. Churg-Strauss syndrome. Allergy 1999; 54: 535-551
- 127 Masi AT, Hunder GG, Lie JT. et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094-1100
- 128 Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol 2012; 24: 24-30
- 129 Guillevin L, Lhote F, Gayraud M. et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17-28
- 130 Nagel MA, White T, Khmeleva N. et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol 2015; 72: 1281-1287
- 131 Borruat FX, Bogousslavsky J, Uffer S. et al. Orbital infarction syndrome. Ophthalmology 1993; 100: 562-568
- 132 Stone JH, Tuckwell K, Dimonaco S. et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med 2017; 377: 317-328
- 133 Seyahi E. Takayasu arteritis: an update. Curr Opin Rheumatol 2017; 29: 51-56
- 134 Tugal-Tutkun I. Systemic vasculitis and the eye. Curr Opin Rheumatol 2017; 29: 24-32
- 135 Misra DP, Wakhlu A, Agarwal V. et al. Recent advances in the management of Takayasu arteritis. Int J Rheum Dis 2019; 22 (Suppl. 01) 60-68
- 136 Iliescu DA, Timaru CM, Batras M. et al. Cogan's Syndrome. Rom J Ophthalmol 2015; 59: 6-13
- 137 Tayer-Shifman OE, Ilan O, Tovi H. et al. Cogan's syndrome--clinical guidelines and novel therapeutic approaches. Clin Rev Allergy Immunol 2014; 47: 65-72
- 138 Grasland A, Pouchot J, Hachulla E. et al. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford) 2004; 43: 1007-1015
- 139 Gluth MB, Baratz KH, Matteson EL. et al. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc 2006; 81: 483-488
- 140 Padoan R, Cazzador D, Pendolino AL. et al. Cogan's syndrome: new therapeutic approaches in the biological era. Expert Opin Biol Ther 2019; 19: 781-788
- 141 Kokturk A. Clinical and Pathological Manifestations with Differential Diagnosis in Behcet's Disease. Patholog Res Int 2012; 2012: 690390
- 142 Hammami S, Yahia SB, Mahjoub S. et al. Orbital inflammation associated with Behcet's disease. Clin Exp Ophthalmol 2006; 34: 188-190
- 143 Urruticoechea-Arana A, Cobo-Ibanez T, Villaverde-Garcia V. et al. Efficacy and safety of biological therapy compared to synthetic immunomodulatory drugs or placebo in the treatment of Behcet's disease associated uveitis: a systematic review. Rheumatol Int 2019; 39: 47-58
- 144 Bettiol A, Silvestri E, Di Scala G. et al. The right place of interleukin-1 inhibitors in the treatment of Behcet's syndrome: a systematic review. Rheumatol Int 2019; 39: 971-990
- 145 Hedrich CM, Schnabel A, Hospach T. Kawasaki Disease. Front Pediatr 2018; 6: 198
- 146 Demirsoy S, Gucuyener K, Olgunturk R. et al. A case of Kawasaki syndrome associated with preseptal cellulitis in orbita. Turk J Pediatr 1988; 30: 55-59
- 147 Felz MW, Patni A, Brooks SE. et al. Periorbital vasculitis complicating Kawasaki syndrome in an infant. Pediatrics 1998; 101: E9
- 148 Sheard RM, Pandey KR, Barnes ND. et al. Kawasaki disease presenting as orbital cellulitis. J Pediatr Ophthalmol Strabismus 2000; 37: 123-125
- 149 Son MB, Gauvreau K, Burns JC. et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr 2011; 158: 644-649 e641
- 150 Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm 2004; 12: 87-99
- 151 Arthurs BP, Khalil MK, Chagnon F. et al. Orbital infarction and melting in a patient with systemic lupus erythematosus. Ophthalmology 1999; 106: 2387-2390
- 152 Nabili S, McCarey DW, Browne B. et al. A case of orbital myositis associated with rheumatoid arthritis. Ann Rheum Dis 2002; 61: 938-939
- 153 Sanchez IJ, Finger DR, Bradshaw DJ. Orbital inflammatory syndrome and systemic lupus erythematosus. J Clin Rheumatol 1995; 1: 295-298
- 154 Kokotis P, Theodossiadis P, Bouros C. et al. Bilateral ocular myositis as a late complication of dermatomyositis. J Rheumatol 2005; 32: 379-381
- 155 Lokdarshi G, Pushker N, Bajaj MS. Sclerosing Lesions of the Orbit: A Review. Middle East Afr J Ophthalmol 2015; 22: 447-451
- 156 Petrarolha SM, Rodrigues BS, Filho FD. et al. Unilateral Eyelid Edema as Initial Sign of Orbital Sarcoidosis. Case Rep Ophthalmol Med 2016; 2016: 6912927
- 157 Rosenbaum JT, Choi D, Wilson DJ. et al. Parallel Gene Expression Changes in Sarcoidosis Involving the Lacrimal Gland, Orbital Tissue, or Blood. JAMA Ophthalmol 2015; 133: 770-777
- 158 Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol 2007; 144: 769-775
- 159 Demirci H, Christianson MD. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol 2011; 151: 1074-1080 e1071
- 160 McNab AA. The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond) 2018; 32: 248-261
- 161 Pasadhika S, Rosenbaum JT. Ocular Sarcoidosis. Clin Chest Med 2015; 36: 669-683
- 162 Aluclu MU, Keklikci U, Guzel A. et al. Melkersson-Rosenthal syndrome with partial oculomotor nerve palsy. Ann Saudi Med 2008; 28: 135-137
- 163 Shapiro M, Peters S, Spinelli HM. Melkersson-Rosenthal syndrome in the periocular area: a review of the literature and case report. Ann Plast Surg 2003; 50: 644-648
- 164 Cocuroccia B, Gubinelli E, Annessi G. et al. Persistent unilateral orbital and eyelid oedema as a manifestation of Melkersson-Rosenthal syndrome. J Eur Acad Dermatol Venereol 2005; 19: 107-111
- 165 Pierre-Filho Pde T, Rocha EM, Natalino R. et al. Upper eyelid oedema in Melkersson-Rosenthal syndrome. Clin Exp Ophthalmol 2004; 32: 439-440
- 166 Khandpur S, Malhotra AK, Khanna N. Melkersson-Rosenthal syndrome with diffuse facial swelling and multiple cranial nerve palsies. J Dermatol 2006; 33: 411-414
- 167 Rawlings NG, Valenzuela AA, Allen LH. et al. Isolated eyelid edema in Melkersson-Rosenthal syndrome: a case series. Eye (Lond) 2012; 26: 163-166
- 168 Ramaswamy B, Singh R, Manusrut M. et al. Sclerosing lipogranuloma of the eyelid: unusual complication following nasal packing in endoscopic sinus surgery. BMJ Case Rep. 2015 2015.
- 169 Witschel H, Geiger K. Paraffin induced sclerosing lipogranuloma of eyelids and anterior orbit following endonasal sinus surgery. Br J Ophthalmol 1994; 78: 61-65
- 170 Herrmann B, Rozot P, Baylac F. et al. [Lipogranuloma of the orbit. Apropos of a case]. J Fr Ophtalmol 1996; 19: 780-784
- 171 AlGhafri L, Galindo-Ferreiro A, Maktabi A. et al. Idiopathic orbital lipogranuloma. Int J Ophthalmol 2017; 10: 494-496
- 172 Jakobiec FA, Rai R, Rashid A. et al. Bilateral Eyelid Pseudoptosis From Lipogranulomas of the Preaponeurotic Fat Pads. Ophthalmic Plast Reconstr Surg 2015; 31: e125-131
- 173 Paik JS, Cho WK, Park GS. et al. Eyelid-associated complications after autogenous fat injection for cosmetic forehead augmentation. BMC Ophthalmol 2013; 13: 32
- 174 Park JY, Kim N. Periorbital Lipogranuloma after Facial Autologous Fat Injection and Its Treatment Outcomes. Korean J Ophthalmol 2016; 30: 10-16
- 175 Cafaro G, Croia C, Argyropoulou OD. et al. One year in review 2019: Sjogren's syndrome. Clin Exp Rheumatol 2019; 37 (Suppl. 118) 3-15
- 176 Mathews PM, Hahn S, Hessen M. et al. Ocular complications of primary Sjogren syndrome in men. Am J Ophthalmol 2015; 160: 447-452 e441
- 177 Danlos FX, Daoued F, Seror R. et al. Orbital Myositis and Primary Sjogren Syndrome. J Rheumatol 2015; 42: 1536-1537
- 178 Tonami H, Matoba M, Kuginuki Y. et al. Clinical and imaging findings of lymphoma in patients with Sjogren syndrome. J Comput Assist Tomogr 2003; 27: 517-524
- 179 Luemsamran P, Rootman J, White VA. et al. The role of biopsy in lacrimal gland inflammation: A clinicopathologic study. Orbit 2017; 36: 411-418
- 180 Ohshima K, Matsuo N, Yokoe S. et al. [A case of lacrimal gland malignant lymphoma, associated with Sjogren's syndrome]. Nippon Ganka Gakkai Zasshi 1991; 95: 386-392
- 181 Carubbi F, Cipriani P, Marrelli A. et al. Efficacy and safety of rituximab treatment in early primary Sjogren's syndrome: a prospective, multi-center, follow-up study. Arthritis Res Ther 2013; 15: R172
- 182 Kimura T. On the unusual granulations combined with hyperplastic changes of lymphatic tissue. Trans Soc Pathol Jpn 1948; 37: 179-180
- 183 Buder K, Ruppert S, Trautmann A. et al. Angiolymphoid hyperplasia with eosinophilia and Kimura's disease - a clinical and histopathological comparison. J Dtsch Dermatol Ges 2014; 12: 224-228
- 184 Claros P, Fokouo V, Nyada F. et al. Kimura's disease of the orbit: A modern diagnostic challenge. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 287-289
- 185 Googe PB, Harris NL, Mihm MC. Kimura's disease and angiolymphoid hyperplasia with eosinophilia: two distinct histopathological entities. J Cutan Pathol 1987; 14: 263-271
- 186 Kodama T, Kawamoto K. Kimura's disease of the lacrimal gland. Acta Ophthalmol Scand 1998; 76: 374-377
- 187 Monzen Y, Kiya K, Nishisaka T. Kimura's Disease of the Orbit Successfully Treated with Radiotherapy Alone: A Case Report. Case Rep Ophthalmol 2014; 5: 87-91
- 188 Bullock JD, Bartley GB, Campbell RJ. et al. Necrobiotic xanthogranuloma with paraproteinemia. Case report and a pathogenetic theory. Ophthalmology 1986; 93: 1233-1236
- 189 Shams PN, Rasmussen SL, Dolman PJ. Adult-Onset Asthma Associated With Simultaneous Conjunctival, Eyelid, and Orbital Xanthogranulomatosis Responsive to Systemic Immunosuppression. Ophthalmic Plast Reconstr Surg 2015; 31: e162-163
- 190 Elner VM, Mintz R, Demirci H. et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthalmic Plast Reconstr Surg 2006; 22: 36-40
- 191 Efebera Y, Blanchard E, Allam C. et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011; 11: 298-302
- 192 Cives M, Simone V, Rizzo FM. et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015; 95: 1-11
- 193 Estrada-Veras JI, O'Brien KJ, Boyd LC. et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 2017; 1: 357-366
- 194 Merritt H, Pfeiffer ML, Richani K. et al. Erdheim-Chester disease with orbital involvement: Case report and ophthalmic literature review. Orbit 2016; 35: 221-226
- 195 Manousaridis K, Casper J, Schittkowski MP. et al. [Erdheim-Chester disease of the orbit with compressive optic neuropathy]. Ophthalmologe 2010; 107: 266-269
- 196 Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D. et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75: 157-169
- 197 Altman J, Winkelmann RK. Diffuse normolipemic plane xanthoma. Generalized xanthelasma. Arch Dermatol 1962; 85: 633-640
- 198 Zak IT, Altinok D, Neilsen SS. et al. Xanthoma disseminatum of the central nervous system and cranium. AJNR Am J Neuroradiol 2006; 27: 919-921
- 199 Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011; 147: 459-464
- 200 Minkov M, Lehrnbecher T. S1-Leitlinie 025/015 „Langerhanszell-Histiozytose (LCH) im Kindesalter In. 2017
- 201 Bertelmann E, Minko N, Lohneis P. et al. Benigne Neoplasien der Orbita. Klin Monatsbl Augenheilkd 2011; 228: 1113-1130
- 202 Maccheron LJ, McNab AA, Elder J. et al. Ocular adnexal Langerhans cell histiocytosis clinical features and management. Orbit 2006; 25: 169-177
- 203 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthalmic Plast Reconstr Surg 2016; 32: 28-34
- 204 Vemuganti GK, Naik MN, Honavar SG. Rosai dorfman disease of the orbit. J Hematol Oncol 2008; 1: 7
- 205 Bothra N, Kaliki S, Gowrishankar S. et al. Isolated unilateral eyelid Rosai-Dorfman disease. Oman J Ophthalmol 2018; 11: 300-302
- 206 Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63-70
- 207 Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19-73
- 208 McClellan SF, Ainbinder DJ. Orbital Rosai-Dorfman disease: a literature review. Orbit 2013; 32: 341-346
- 209 Ezeanosike E, Ezeanosike OB, Akpan SI. et al. Rhabdomyoblastic differentiation in rosai dorfman disease of the orbit in a 12-year-old male. Niger J Clin Pract 2018; 21: 1670-1673
- 210 Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123: 2924-2933
- 211 Goel R, Raut A, Agarwal A. et al. A Rare Presentation of Orbital Castleman's Disease. Case Rep Ophthalmol Med 2020; 2020: 1012759
- 212 Schrock A, Gutgemann I, Keiner S. [Castleman's disease in ear, nose, and throat practice]. HNO 2007; 55 (Suppl. 01) E29-32
- 213 Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: a systematic review of 404 published cases. Ann Surg 2012; 255: 677-684
- 214 van Rhee F, Wong RS, Munshi N. et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014; 15: 966-974
- 215 Kesper C, Viestenz A, Siebolts U. et al. [Orbital Amyloidosis - Comparison of Two Different Clinical Courses]. Klin Monbl Augenheilkd 2020; 237: 35-40
- 216 Leibovitch I, Selva D, Goldberg RA. et al. Periocular and orbital amyloidosis: clinical characteristics, management, and outcome. Ophthalmology 2006; 113: 1657-1664
- 217 Mora-Horna ER, Rojas-Padilla R, Lopez VG. et al. Ocular adnexal and orbital amyloidosis: a case series and literature review. Int Ophthalmol 2016; 36: 281-298
- 218 Naxer S, Behnes CL, Schittkowski MP. [Amyloidosis--a rare differential diagnosis of an orbital tumour]. Klin Monbl Augenheilkd 2011; 228: 555-564
- 219 Ladner-Merz S, Muller-Ladner U. [Amyloidoses]. Z Rheumatol 2008; 67: 677-682 quiz 683
- 220 Khaira M, Mutamba A, Meligonis G. et al. The use of radiotherapy for the treatment of localized orbital amyloidosis. Orbit 2008; 27: 432-437
- 221 Eghbali A, Hassan S, Seehra G. et al. Ophthalmological findings in Gaucher disease. Mol Genet Metab 2019; 127: 23-27
- 222 Prakalapakorn SG, Proia AD, Yanovitch TL. et al. Ocular and histologic findings in a series of children with infantile pompe disease treated with enzyme replacement therapy. J Pediatr Ophthalmol Strabismus 2014; 51: 355-362
- 223 Slingerland NW, Polling JR, van Gelder CM. et al. Ptosis, extraocular motility disorder, and myopia as features of pompe disease. Orbit 2011; 30: 111-113
- 224 Winter AW, Salimi A, Ospina LH. et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol 2019; 103: 315-326
- 225 Yanovitch TL, Banugaria SG, Proia AD. et al. Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 2010; 47: 34-40
- 226 Wabbels B, Ali N, Kunz WS. et al. [Chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome : interdisciplinary diagnosis and therapy]. Ophthalmologe 2008; 105: 550-556
- 227 Pfeiffer MJ. [Chronic Progressive External Ophthalmoplegia Ptosis: Problems with Diagnostics and Treatment]. Klin Monbl Augenheilkd 2018; 235: 31-33
- 228 Pitceathly RD, Morrow JM, Sinclair CD. et al. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol 2016; 26: 130-137
- 229 Rahman S, Blok RB, Dahl HH. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996; 39: 343-351
- 230 McKelvie P, Infeld B, Marotta R. et al. Late-adult onset Leigh syndrome. J Clin Neurosci 2012; 19: 195-202
- 231 Lake NJ, Compton AG, Rahman S. et al. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 2016; 79: 190-203
- 232 Han J, Lee YM, Kim SM. et al. Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol 2015; 99: 528-535
- 233 Fichter N, Schittkowski M, Guthoff R. Erkrankungen der Tränendrüse. Ophthalmologe 2005; 102: 399-425
- 234 Moreiras J. Tumors of the lacrimal gland - Orbit – examination, diagnosis, mircrosurgery, pathology. 2004
- 235 Font R, Gamel J. Epithelial tumours of the lacrimal gland: an analysis of 265 cases.In Jakobiec F (ed) Ocular and adnexal tumours. Aesculapius, Birmingham. 1978
- 236 Riedel K, Markl A, Hasenfratz G. et al. Epithelial tumors of the lacrimal gland: Clinico-pathologic correlation and management. Neurosurg Rev 1990; 13: 289-298
- 237 Andreasen S, Esmaeli B, Holstein L. et al. An Update on Tumors of the Lacrimal Gland. Asia-Pac. J Ophthalmol 2017; 6: 159-172
- 238 Cates C, Manners R, Rose G. Pleomorphic adenoma of the lacrimal gland in a 10 year old girl. Br J Ophthalmol 2002; 86: 249-250
- 239 Faktorovich E, Crawford J, Char D. Benign mixed tumor (pleomorphic adenoma) of the lacrimal gland in a 6-year-old boy. Am J Ophthalmol 1996; 122: 446-447
- 240 Mercado G, Gunduz K, Shields C. et al. Pleomorphic adenoma of the lacrimal gland in a teenager. Arch Ophthalmol 1998; 116: 962-963
- 241 Ni C, Kuo P, Dryja T. Histopathological classification of 272 primary epithelial tumors of the lacrimal gland. Chin Med J 1992; 105: 481-485
- 242 Rose G, Wright J. Pleomorphic adenoma of the lacrimal gland. Br J Ophthalmol 1992; 76: 395-400
- 243 Stefko S, DiBernardo C, Green W. et al. Pleomorphic adenoma of the lacrimal gland with extensive calcification. Arch Ophthalmol 2004; 122: 778-780
- 244 Stupp T, Pavlidis M, Buchner T. Pleomorphic Adenoma of the Lacrimal Gland in a Child After Treatmentof Acute Lymphoblastic Leukemia. Arch Ophthalmol 2004; 122: 1538-1540
- 245 Bonavolontà G, Tranfa F, Staibano S. et al. Warthin Tumor of the Lacrimal Gland. Am J Ophthalmol 1997; 124: 857-858
- 246 Ghosh D, Ekka J. Adenolymphoma of the lacrimal gland: A very rare case report. Int J Ocul Oncol oculoplasty 2018; 4: 144-145
- 247 Bartoletti-Stella A, Salfi N, Ceccarelli C. et al. Mitochondrial DNA Mutations in Oncocytic Adnexal Lacrimal Glands of the Conjunctiva. ARCH OPHTHALMOL 2011; 129: 664-666
- 248 Calle C, Castillo I, Eagle R. et al. Oncocytoma of the Lacrimal Gland: Case Report and Review of the Literature. Orbit 2006; 25: 243-247
- 249 Cameron J, Barras C. Oncocytoma of the eyelid. ACTA OPHTHALMOL SCAND 2004; 83: 125
- 250 George E, Swanson P, Newman B. et al. Oculocutaneous oncocytic tumors: clinicopathologic and immunohistochemical study of 2 cases with literature review. Am J Dermatopathol 2007; 29: 279-285
- 251 Morand B, Bettega G, Bland V. et al. Oncocytoma of the eyelid: an aggressive benign tumor. Ophthalmology 1998; 105: 2220-2224
- 252 Shields C, Shields J. Tumors of the conjunctiva and cornea. Survey of Ophthalmology 2004; 49: 3-24
- 253 Shields C, Shields J, Arbizo V. Oncocytoma of the caruncle. Am J Ophthalmol 1986; 102: 315-319
- 254 Surakiatchanukul T, Sioufi K, Pointdujour-Lim R. et al. Caruncular Oncocytoma Mimicking Malignant Melanoma. Ocul Oncol Pathol 3: 320-323
- 255 Bolzoni A, Pianta L, Farina D. et al. Benign myoepithelioma of the lacrimal gland: report of a case. Eur Arch Otorhinolaryngol 2005; 262: 186-188
- 256 Font R, Garner A. Myoepithelioma of the lacrimal gland: report of a case with spindle cell morphology. Brit J Ophthalmol 1992; 76: 634-636
- 257 Grossniklaus H, Wojno T, Wilson M. et al. Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1997; 115: 1588-1590
- 258 Heathcote J, Hurwitz J, Dardick I. A Spindle-Cell Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1990; 108: 1135-1139
- 259 Nickol T, E R Kleinsasser O. Myoepitheliome der Parotis. Jahresversammlung der Deutschen Gesellschaft für HNO-Heilkunde. 1998
- 260 Bajaj M, Pushker N, Kashyap S. Cystadenoma of the lacrimal gland. Orbit 2002; 21: 301-305
- 261 Bernardini F, Devoto M. JO C. Epithelial tumors of the lacimal gland: an update. Curr Opin in Ophthalmol 2008; 19: 409-413
- 262 Silbert J, Braich P, Misra R. Basal cell cystadenoma of the lacrimal gland: diagnostic pitfalls of a basaloid pattern in lacrimal tumours. Internat Ophthalmol. 2011 DOI: 43-46
- 263 Wang L, Zhang SK, Ma Y. et al. Papillary cystadenoma of the parotid gland: A case report. World J Clin Cases 2019; 7: 366-372
- 264 Pfeiffer M, Yin V, Bell D. et al. Sclerosing Polycystic Adenosis of the Lacrimal Gland. Ophthalmology 2013; 120: 873
- 265 Speight P, Barrett A. Salivary gland tumours: diagnostic challenges and an update on the latest WHO classification. Diagn Histopathol 2020; 26: 147-158
- 266 Brunnmann R, Ro J, Ordonez N. et al. Extrapleural Solitary Fibrous Tumor: A Clinicopathologic Study of 24 Cases. Mod Pathol 1999; 12: 1034-1042
- 267 Mupas-Uy J, Kitaguchia Y, Takahashia Y. et al. Solitary Fibrous Tumor in the Lacrimal Gland Fossa: A Case Report. Case Rep Ophthalmol 2016; 7: 398-403
- 268 Okike N, Bernatz P, Woolner L. Localized mesothelioma of the pleura. Benign and malignant variants. J Thorac Cardiovasc Surg 1978; 75: 363-372
- 269 Scott I, Tanenbaum M, Rubin D. Solitary fibrous tumor of the lacrimal gland fossa. Ophthalmology 1996; 103: 1613-1618
- 270 Vallat-Decouvelaere A, Dry S, Fletcher C. Atypical and malignant solitary fibrous tumor in extrathoracic locations; evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol 1998; 22: 1501-1511
- 271 Woo K, Suh Y, Kim Y. Solitary fibrous tumor of the lacrimal sac. Ophthalmic Plast Reconstr Surg 1999; 15: 450-453
- 272 Bielory BP, Turbin RE, Mirani NM. et al. Radiographic and histological findings in an atypical orbital myxoma. Ophthalmol Eye Dis 2011; 3: 55
- 273 Demirci H, Shields CL, Eagle RCJ. et al. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006; 124: 735-738
- 274 Kini Rao A, Nayal B. Conjunctival myxoma-a case report. Malays J Med Sci 2013; 20: 92-94
- 275 Parikh D, Mukherjee B. Lacrimal gland myxoma. Indian J Ophthalmol 2017; 65: 887-889
- 276 Rambhatla S, Subramanian N, Gangadhara Sundar JK. et al. Myxoma of the orbit. Indian J Ophthalmol 2003; 51: 85-87
- 277 Bajaj M, Pushker N, Fibrous SK. histiocytoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 145-147
- 278 Andrew N, Coupland S, Pirbhai A. et al. Lymphoid hyperplasia of the orbit and ocular adnexa: A clinical pathologic review. Survey of Ophthalmology 2016; 61: 778-790
- 279 Shields J, Shields C, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: the 2002 Montgomery lecture, part 1. Ophtalmology. 2004 DOI: 997-1008
- 280 Wittekind C. TNM-Klassifikation maligner Tumoren. Achte Auflage. Aufl. WILEY-VCH; 2017
- 281 Devoto M, Croxatto J. Primary cystadenocarcinoma of the lacrimal gland. Ophthalmology 2003; 110: 2006-2010
- 282 Takahira M, Nakamura Y, Shimizu S. et al. Carcinosarcoma of the Lacrimal Gland Arising From a Pleomorphic Adenoma. Am J Ophthalmol 2005; 140: 340-341
- 283 Daisuke Y, Toshiyuki O, Shuichi Y. Case of primary neuroendocrine carcinoma in lacrimal gland. Acta Ophthalmol 2013; 91: 252
- 284 Gess A, Silkiss R. A merkel cell carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 28. 11-13
- 285 El-Naggar A, Chan J, Grandis J. World Health organization classification of head and neck tumours. 2017 DOI:
- 286 Weis E, Rootman J, Joly T. et al. Epithelial lacrimal gland tumors: pathologic classification and current understanding. Arch Ophthalmol 2009; 127: 1016-1028
- 287 Gamel J, Font R. Adenoidcystic carcinoma of the lacrimal gland: the clinical significance of a basaoid histologic pattern. Human Pathol 1982; 13: 219-225
- 288 Friedrich R, Bleckmann V. Adenoid cystic carcinoma of the salivary and lacrimal glands origin: localization, classification, clinical pathological correlation, treatment results and long-term follow-up control in 84 patients. Anticancer Res 2003; 23: 937-940
- 289 Goldberg R. Intraarterial chemotherapy. A welcome new idea for the management of adenoidcystic carcinoma of the lacrimal gland. Arch Ophthalmol 1998; 116: 372-373
- 290 Hamper K, Lazar F, Dietel M. Prognostic factors for adenoid cystic carcinoma of the head and neck: a retrospectiv evaluation of 96 cases. J Oral Pathol Med 1990; 19: 101-107
- 291 Henderson J. In Orbital Tumors New York: Raven Press;. 1994: 323-342
- 292 Lee D, Campbell R, Waller R. A clinicopathologic study of primary adenoid cystic carcinoma of the lacrimal gland. Ophthalmology 1985; 92: 128-134
- 293 Strianese D, Baldi G, Staibano S. Expression of apoptosis-related markers in malignant epithelial tumors of the lacrimal gland and their relation to clinical outcome. Br J Ophthalmol 2007; 91: 1239-1243
- 294 Tse D, Benedetto P, Dubovy S. Clinical analysis oft he effect on intraarterial cytoreductive chemotherapy in the treatment of lacrimal gland andoid cystic carcinoma. Am J Ophthalmol 2006; 141: 44-53
- 295 Wright J, Rose G, Garner A. Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 1992; 76: 401-407
- 296 Karcioglu Z. Orbital Tumors. Diagnosis and Management. New York: Springer; 2005
- 297 Ahmad S, Esmaeli B, Williams M. American Joint Committee on Cancer classification predicts outcome of patients with lacrimal gland adenoid cystic carcinoma. Ophthalmology 2009; 116: 1210-1215
- 298 GROUP IHANS. Cervical lymph node metastasis in adenoid cystic carcinoma of the sinonasal tract, nasopharynx, lacrimal glands and external auditory canal: a collective international review. J Laryngol Otol 2016; 130: 1093-1097
- 299 Xiao R, Sethi R, Feng A. et al. The role of elective neck dissection in patients with adenoid cystic carcinoma of the head and neck. Laryngoscope 2019; 129: 2094-2104
- 300 Bördlein I. Mit schweren Ionen gegen hartnäckige Tumoren. Deutsches Ärzteblatt 2004; 101: 1718-1720
- 301 Hayashi K, Koto M, Ikawa H. et al. Efficacy and safety of carbon-ion radiotherapy for lacrimal gland carcinomas with extraorbital extension: a retrospective cohort study. Oncotarget 2018; 9: 12932-12940
- 302 Lesueur P, Rapeaud E, De Marzi L. et al. Adenoid Cystic Carcinoma of the Lacrimal Gland: High Dose Adjuvant Proton Therapy to Improve Patients Outcomes. Front Oncol 2020; 10: 135
- 303 Wolkow N, Jakobiec F, Lee H. et al. Long-term Outcomes of Globe-Preserving Surgery With Proton Beam Radiation for Adenoid Cystic Carcinoma of the Lacrimal Gland. Am J Ophthalmol. 2018 195.
- 304 Shields J, Shields C, Freire J. Plaque radiotherapy for selected orbital malignancies: preliminary observations: hte 2002 Montgomery Lecture, part 2. Ophthal Plast Reconstr Surg 2003; 19: 91-95
- 305 Tyl J, Blank L, Koornneef L. Brachytherapy in orbital tumors. Ophthalmology 1997; 104: 1475-1479
- 306 Gündüz A, Yesiltas Y, Shileds C. Overview of benign and malignant lacrimal gland tumors. Curr Opin Ophthalmol 2018; 29: 458-468
- 307 Ashton N. Epithelial tumors of the lacrimal gland. Mod Probl Ophthalmol 1975; 14: 306-323
- 308 Perzin K, Jakobiec F, Lovolsi V. Lacrimal gland malignant mixed tumors (carcinomas arising in benign mixed tumors): a clinicopathologic study. Cancer 1980; 45: 2593-2606
- 309 Shields C, Shields J, Eagle R. Clinicopathologic review of 142 cases of lacrimal gland lesions. Ophthalmology 1989; 96: 431-435
- 310 Henderson J, Farrow G. Primary malignant mixed tumors of the lacrimal gland. Report of 10 cases. Ophthalmology 1980; 87: 466-475
- 311 Jakobiec F, Bilyk J, Font R. Ophthalmic Pathology. An Atlas and Textbook. 4th ed. Aufl. Philadelphia. WB Saunders; 1996
- 312 Chen A, Garcia J, Bucci M. et al. The role of postoperative radiation therapy in carcinoma ex pleomorphic adenoma of the parotid gland. Int J Radiat Oncol Biol Phys 2007; 67: 138-143
- 313 Takahira M, Minato H, Takahashi M. Cystic carcinoma ex pleomorphic adenoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 407-409
- 314 Hwang S, Kim K. High-grade Mucoepidermoid Carci- noma of the Lacrimal Gland. Korean J Ophthalmol 2018; 32: 426-427
- 315 Heaps R, Miller N, Albert D. Primary adenocarcinoma of the lacrimal gland. Ophtalmology 1993; 100: 1856-1860
- 316 von Holstein S, Coupland S. , D B. Epithelial Tumors of the Lacrimal Gland: a clinical, histopathological, surgical, and oncological survey. Acta Ophthalmol 2013; 91: 195-206
- 317 Dennie T. Metastastic her-2 amplified lacrimal gland carcinoma with response to lapatinib treatment. Case Rep Oncol Med 2015; 2015: 1-3
- 318 Totuk O, Demir M, Yapicier O. et al. Low-Grade Mucoepidermoid Carcinoma of the Lacrimal Gland in a Teenaged Patient. Case Rep Ophthalmol Med. 2017
- 319 Eviatar J, Hornblass A. Mucoepidermoid carcinoma of the lacrimal gland: 25 cases and a review and update of the literature. Ophthal Plast Reconstr Surg 1993; 9: 170-181
- 320 Sofinski S, Brown B, Rao N. Mucoepidermoid carcinoma of the lacrimal gland. Case report and review of the literature. Ophthalmic Plast Reconstr Surg 1986; 2: 147-151
- 321 Thorvaldsson S, Beahrs O, Woolner L. et al. Mucoepidermoid tumors of the major salivary glands. Am J Surg 1970; 120: 432-438
- 322 Katz E, Rootman J, Dolman P. et al. Primary Ductal Adenocarcinoma of the Lacrimal Gland. Ophthalmology 1996; 103: 157-162
- 323 Yang H, Wu C, Tsai C. et al. Primary ductal adenocarcinoma of lacrimal gland: Two case reports and review of the literature. Taiwan J Ophthalmol 2018; 8: 42-48
- 324 Ishida M, Hotta M, Kushima R. et al. Case of ductal adenocarcinoma ex pleomorphic adenoma of the lacrimal gland. Rinsho Byori 2009; 57: 746-751
- 325 Andreasen S, Grauslund M, Heegaard S. Lacrimal gland ductal carcinomas: clinical, morphological and genetic characterization and implications for targeted treatment. Acta Ophthalmol 2017; 95: 299-306
- 326 Jaspers H, Verbist B, Schoffelen R. et al. Androgen Receptor–Positive Salivary Duct Carcinoma: A Disease Entity With Promising New Treatment Options. J Clin Oncol 2011; 29: 473-476
- 327 See T, Stålhammar G, Tang T. et al. Primary ductal adenocarcinoma of the lacrimal gland: A review and report of five cases. Survey of Ophthalmology 2020; 65: 371-380
- 328 Anesi A, Negrello S, Lucchetti D. et al. Clinical Management of Acinic Cell Carcinoma of the Lacrimal Gland. Case Rep Oncol 2019; 12: 777-790
- 329 De Rosa G, Zeppa P, Tranta F. Acinic Cell Carcinoma in a Lacrimal Gland: first case report. Cancer 1986; 57: 1988-1991
- 330 Jang J, Kie J, Lee S. Acinic cell carcinoma of the lacrimal gland with intracranial extension. Ophthalmic Plast Reconstr Surg 2001; 17: 454-457
- 331 Rosenbaum P, Mahadevia P, Goodman L. Acinic cell carcinoma of the lacrimal gland. Arch Ophthalmol 1995; 113: 781-785
- 332 Briscoe D, Mahmood S, Bonshek R. et al. Primary sebaceous carcinoma of the lacrimal gland. Br J Ophthalmol 2001; 85: 625-626
- 333 Harvey P, Parsons M, Rennie I. Primary sebaceous carcinoma of lacrimal gland: a previously unreported primary neoplasm. Eye 1994; 8: 592-595
- 334 Kiratli H, Tarlan B, Fırat P. Primary sebaceous carcinoma of the lacrimal gland. Orbit 2012; 31: 352-354
- 335 Park H, Choi S. Primary sebaceous carcinoma of lacrimal gland: A case report and review of literature. World J Clin Cases 2018; 6: 1194-1198
- 336 Yamamoto N, Mizoe J, Hasegawa A. et al. Primary sebaceous carcinoma of the lacrimal gland treated by carbon ion radiotherapy. Int J Clin Oncol 2003; 8: 386-390
- 337 Herrera G. Light microscopic, ultrastructural and immunocytochemical spectrum of malignant lacrimal and salsivary gland tumors, including malignant mixed tumors. Pathobiology 1990; 58: 312-322
- 338 Iida K, Shikishima K, Okido M. et al. A case of malignant myoepithelioma in the lacrimal gland. Nippon Ganka Gakkai Zasshi 2001; 105: 42-46
- 339 Larbcharoensub N, Pangpunyakulchai D, Aroonroch R. et al. Lacrimal myoepithelial carcinoma ex recurrent pleomorphic adenoma: A clinicopathological report and review of the literature. Mol Clin Oncol 2018; 8: 209-213
- 340 Nagao T, Sugano I, Ishida Y. Salivary gland mmalignant myoepithelioma: a clinicopathologic and immunohistochemical study of ten cases. Cancer 1998; 83: 1292-1299
- 341 Xu T, Liao Z, Tang J. et al. Myoepithelial carcinoma of the headand neck: A report of 23 casesand literature review. Cancer Treatment. Communications 2014; 2: 24-29
- 342 Mahdi Y, Azami M, Daoudi R. et al. Diagnostic pitfall: primary myoepithelial carcinoma of the lacrimal gland, case report and literature review. BMC Clinical Pathology 2018; 18: 1-7
- 343 Yu G, Ma D, Sun K. Myoepithelial carcinoma of the salivary glands: behavior and management. Chin Med J 2003; 116: 163-165
- 344 Soumarová R, Lovasová Z, Vaňková M. Radiotherapy in malignant myoepithelioma of the soft palate – case report. Case Rep Oncol 2009; 2: 116-120
- 345 Nieder C, Schneller F, Grosu A. Radiotherapy and chemotherapy for myoepithelioma of the sellar region. Strahlenther Onkol 2005; 181: 260-263
- 346 Fenton S, Srinivasan S, Harnett A. et al. Primary squamous cell carcinoma of the lacrimal gland. Eye 2003; 17: 424-425
- 347 Su G, Patipa M, Font R. Primary squamous cell carcinoma arising from an epithelium-lined cyst of the lacrimal gland. Ophthal Plast Reconstr Surg 2005; 21: 383-385
- 348 Hotta K, Arisawa T, Mito H. et al. Primary squamous cell carcinoma of the lacrimal gland. Clin Experiment Ophthalmol 2005; 33: 534-536
- 349 Bernardini F, Orcioni G, Croxatto J. Oncocytic carcinoma of the lacrimal gland in a patient with neurofibromatosis. Ophthal Plast Reconstr Surg 2010; 26: 486-488
- 350 Kalantzis G, Norris J, El-Hindy N. et al. Oncocytic adenocarcinoma of the lacrimal gland: an unusual presentation. Eye 2013; 27: 104-105
- 351 Selva D, Davis G, Dodd T. Polymorphous Low-Grade Adenocarcinoma of the Lacrimal Gland. ARCH OPHTHALMOL 2004; 122: 915-917
- 352 Bortz J, Zhang P, Eagle R. et al. Secretory Carcinoma of the Lacrimal Gland: A Rare Case Report. Ophthalmic Plast Reconstr Surg 2018; 34: 154-157
- 353 Hyrczaa M, Andreasen S, Melchiorc L. et al. Primary Secretory Carcinoma of the Lacrimal Gland: Report of a New Entity. Am J Ophthalmol 2018; 193: 178-183
- 354 Kadir S. Synovial Sarcoma involving lacrimal gland: A rare clinical entity. Int J Ocu Oncol Oculoplast. 1: 14–18
- 355 Likhvantseva V, Safonova T, Kuzmin K. Primary granulocytic sarcoma of lacrimal gland. Vestn Oftalmol 2016; 132: 82-89
- 356 Yilmaz A, Saydam G, Sahin F. et al. Granulocytic sarcoma: a systematic review. Am J Blood Res 2013; 3: 265-270
- 357 Rajabi M, Riazi H, Hosseini S. et al. Malignant Peripheral Nerve Sheath Tumor of Lacrimal Nerve: A Case Report. Orbit 2015; 34: 41-44
- 358 Stark A, Hugo H, Buhl R. et al. Tumoren peripherer Nerven. Dtsch Arztebl 2002; 99: 928-933
- 359 Gündüz K, Shields J, Eagle R.. Malignant Rhabdoid Tumor of the Orbit. Arch Ophthalmol 1998; 116: 243-246
- 360 Johnson L, Sexton F, Goldberg S. Poorly differentiated primary orbital sarcoma (presumed malignant rhabdoid tumor): radiologic and histopathologic correlation. Arch Ophthalmol 1991; 109: 1275-1278
- 361 Orphanet. Rhabdoidtumor. In: Orphanet; 2020
- 362 Rootman J, Damji K, Dimmick J. Malignant rhabdoid tumor of the orbit. Ophthalmology 1989; 96: 1650-1654
- 363 Walford N, Deferrai R, Slater R. Intraorbital rhabdoid tumour following bilateral retinoblastoma. Histopathology 1992; 20: 170-173
- 364 Niffenegger J, Jakobiec F, Shore J. et al. Adult extrarenal rhabdoid tumor of the lacrimal gland. Ophthalmology 1992; 99: 567-574
- 365 Ferry J, Fung C, Zukerberg L. et al. Lymphoma of the ocular adnexa: A study of 353 cases 2007; 31: 170-184
- 366 Rasmussen P, Ralfkiaer E, Prause J. Malignant lymphoma of the lacrimal gland. A nation-based study. Arch Ophthalmol 2011; 129: 1275-1280
- 367 Johnson T, Tse D, Byrne G. Ocular adnexal lymphoid tumors: a clinicopathologic and molecular genetic study of 77 patients. Ophthal Plast Reconstr Surg 1999; 15: 171-179
- 368 Polito E, Galieni P, Leccisotti A. Clinical and radiological presentation of 95 orbital lymphoid tumors. Graefes Arch Clin Exp Ophthalmol 1996; 234: 504-509
- 369 Aranow M. Ocular adnexal lymphoma: evidence-based treatment approach. Int Ophthalmol Clin 2015; 55: 97-109
- 370 König L, Stade R, Rieber J. Radiotherapy of indolent orbital lymphomas: two radiation concepts. Strahlenther Onkol 2016; 192: 414-421
- 371 de Leval L, Jaffe E. Lymphoma Classification. Cancer J 2020; 26: 176-185
- 372 Shields J, Shields C, Eagle R. Metastatic renal cell carcinoma to the palpebral lobe of the lacrimal gland. Ophthal Plast Reconstr Surg 2001; 17: 191-194
- 373 Fowler J, Terplan K. Fibroma of the Orbit. Arch Ophthalmol 1942; 28: 263-271
- 374 Mortada A. Fibroma of the orbit. Brit J Ophthal 1971; 55: 350
- 375 Rong A, Ulloa-Padilla J, Blessing N. et al. Subperiosteal fibroma of the orbit. Orbit 2018; 37: 378-380
- 376 Rabelo N, da Silva V, do Espírito Santo M. et al. Orbit ossifying fibroma – Case report and literature review. Surg Neurol Int 2020; 11: 35
- 377 Sheikh BY. Cranial aneurysmal bone cyst “with special emphasis on endovascular management”. Acta Neurochir (Wien) 1999; 141: 601-610 discussion 610-601
- 378 Glien A, Meyer B, Wernert N. et al. [Orbital aneurysmal bone cyst in a 2-year-old child]. HNO 2007; 55: 281-286
- 379 Wang JC, Zhang M, Zhao XX. Aneurysmal bone cyst of the orbit. Chin Med J (Engl) 2015; 128: 562-563
- 380 Yu JW, Kim KU, Kim SJ. et al. Aneurysmal bone cyst of the orbit : a case report with literature review. J Korean Neurosurg Soc 2012; 51: 113-116
- 381 Ruiter DJ, van Rijssel TG, van der Velde EA. Aneurysmal bone cysts: a clinicopathological study of 105 cases. Cancer 1977; 39: 2231-2239
- 382 Biesecker JL, Marcove RC, Huvos AG. et al. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer 1970; 26: 615-625
- 383 Schajowicz F. Tumors and tumorlike lesions of bone 2. Aufl. Berlin-Heidelberg-New York: Springer-Verlag; 1994
- 384 Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45: 1-11
- 385 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 314
- 386 Mallen JR, Caranfa JT, Zimmerman D. et al. Atypical presentation of a solid-variant orbital aneurysmal bone cyst with a literature review. Neurochirurgie 2018; 64: 431-433
- 387 Chawla B, Khurana S, Kashyap S. Giant cell reparative granuloma of the orbit. Ophthalmic Plast Reconstr Surg 2013; 29: e94-95
- 388 Kamoshima Y, Sawamura Y, Imai T. et al. Giant cell tumor of the frontal bone in a girl: case report. Neurol Med Chir (Tokyo) 2011; 51: 798-800
- 389 Smolarz-Wojnowska A, Essig H, Gellrich NC. et al. [Orbital tumours in children and adolescents. Differential diagnostics and clinical symptoms]. Ophthalmologe 2010; 107: 543-548
- 390 Vernet O, Ducrey N, Deruaz JP. et al. Giant cell tumor of the orbit. Neurosurgery 1993; 32: 848-851
- 391 Yip CM, Lee HP, Hsu SS. et al. Left orbital roof giant cell tumor of bone: A case report. Surg Neurol Int 2018; 9: 127
- 392 Yamaguchi T, Dorfman HD. Giant cell reparative granuloma: a comparative clinicopathologic study of lesions in gnathic and extragnathic sites. Int J Surg Pathol 2001; 9: 189-200
- 393 de Lange J, van den Akker HP, van den Berg H. Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 104: 603-615
- 394 Auclair PL, Cuenin P, Kratochvil FJ. et al. A clinical and histomorphologic comparison of the central giant cell granuloma and the giant cell tumor. Oral Surg Oral Med Oral Pathol 1988; 66: 197-208
- 395 de Lange J, van Rijn RR, van den Berg H. et al. Regression of central giant cell granuloma by a combination of imatinib and interferon: a case report. Br J Oral Maxillofac Surg 2009; 47: 59-61
- 396 Roman-Romero L, Gonzalez-Garcia R. Cholesterol granuloma of the orbit. Report of cases and analysis of controversial treatment. J Maxillofac Oral Surg 2011; 10: 166-169
- 397 Loeffler KU, Kommerell G. Cholesterol granuloma of the orbit--pathogenesis and surgical management. Int Ophthalmol 1997; 21: 93-98
- 398 Selva D, Phipps SE, O'Connell JX. et al. Pathogenesis of orbital cholesterol granuloma. Clin Exp Ophthalmol 2003; 31: 78-82
- 399 Daus W, Voges J, Schwechheimer K. et al. [Cholesterol granuloma of the orbits--clinicopathologic study]. Klin Monbl Augenheilkd 1988; 193: 195-199
- 400 Hsu HT, Liao WC, Wu CC. et al. Cholesterol granuloma of the orbit. Kaohsiung J Med Sci 2017; 33: 422-423
- 401 Rong AJ, Erickson BP, Blessing NW. et al. Orbital cholesterol granuloma: A report and discussion of orbital findings. Am J Ophthalmol Case Rep 2019; 15: 100468
- 402 Arat YO, Chaudhry IA, Boniuk M. Orbitofrontal cholesterol granuloma: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2003; 19: 382-387
- 403 Aferzon M, Millman B, O'Donnell TR. et al. Cholesterol granuloma of the frontal bone. Otolaryngol Head Neck Surg 2002; 127: 578-581
- 404 Kabra R, Patel S, Shanbhag S. Orbital Chondroma: A rare mesenchymal tumor of orbit. Indian J Ophthalmol 2015; 63: 551-554
- 405 Garrity J, Henderson J. Henderson’s Orbital Tumors.In 4th ed. Aufl. USA: Lippincott Williams and Wilkis; 2007: 103–104
- 406 Rootman J, Connell J. Primary bone tumors of orbit. Surv Ophthalmol 2004; 49: 328-342
- 407 Shields J, Bakewell B, Augsburger J. et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984; 102: 1606-1611
- 408 Pushker N, Meel R, Sharma S. et al. Giant orbital Schwannoma with fluid-fluid-levels. Br J Ophthalmol 2010; 95: 1168
- 409 Misra S, Gogri P, Misra N. et al. Recurrent neurofibroma of the orbit. Australas Med J 2013; 30: 189-191
- 410 Lee L, Gigantelli J, Kincaid M. Localized neurofibroma of the orbit: a radiographic and histopathologic study. Ophthal Plast Reconstr Surg 2000; 16: 241-246
- 411 Turbin R, Thompson C, Kennerdell J. et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophtalmology 2002; 109: 890-899
- 412 Schittkowski M, Fichter N., Orbitale Neoplasien Teil I. Einführung, vaskuläre und neurogene Neoplasien. Ophthalmologe 2012; 109: 1033-1046
- 413 Jain D, Ebrahimi K, Miller N. et al. Intraorbital meningiomas: a pathologic review using current World Health Organization criteria. Arch Pathol Lab Med 2010; 134: 766-770
- 414 Eddleman C, Liu J. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus 2007; 23: E4
- 415 Saeed P, Rootman J, Nugent R. et al. Optic nerve sheath meningiomas. Ophthalmology 2003; 110: 2019-2030
- 416 Ramey W, Arnold S, Chiu A. et al. A Rare Case of Optic Nerve Schwannoma: Case Report and Review of the Literature. Cureus 2015; 7: e265
- 417 Gnekow A. Low-grade chiasmatic-hypothalamic glioma-carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy - report from the multicenter treatment study for children and adolescents with a low grade glimoa - HIT-LGG 1996 - of the Society of Paediatric Oncology and Hematology (GPOH). Klin Padiatr 2004; 216: 331-342
- 418 Sharif S. Second primary tumors in neurofibromatosis 1 patients treated for optic nerve glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24: 2570-2575
- 419 Wilhelm H. Primary optic nerve tumors. Curr Opin Neurol 2009; 22: 11-18
- 420 Emesz M, Arlt E, Krall E. et al. Granularzelltumor der Orbita. Der. Ophthalmologe 2014; 111: 866-870
- 421 Huang N, Rayess H, Svider P. et al. Orbital Paraganglioma: A Systematic Review. J Neurol Surg B Skull Base 2017; 79: 407-412
- 422 Makhdoomi R, Nayil K, Santosh V. et al. Orbital paraganglioma--a case report and review of the literature. Clin Neuropathol 2010; 29: 100-104
- 423 Al-Khiary H, Ayoubi A, Elkhamary S. Primary orbital ganglioneuroma in a 2-year-old healthy boy. Saudi J Ophthalmol 2010; 24: 101-104
- 424 Cannon T, Brown H, Hughes B. Orbital Ganglioneuroma in a Patient With Chronic Progressive Proptosis. Arch Ophthalmol 2004; 122: 1712-1714
- 425 Hatsukawa Y, Furukawa A, Kawamura H. et al. Rhabdomyoma of the orbit in a child. Am J Ophthalmol 1997; 123: 142-144
- 426 Li Y, Li N, Zang W. et al. Rhabdomyoma of the orbit. J Pediatr Ophthalmol Strabismus 2008; 45: 113-115
- 427 Myung J, Kim I, Chun J et al. Rhabdomyoma of the orbit: a case report. Pediatric Radiolog. y. 32: 589–592
- 428 Gündüz K, Günalp I, Erden E. et al. Orbital leiomyoma: report of a case and review of the literature. Surv Ophthalmol 2004; 49: 237-242
- 429 Merani R, Khannah G, Mann S. et al. Orbital leiomyoma: a case report with clinical, radiological and pathological correlation. Clin Exp Ophthalmol 2005; 33: 408-411
- 430 Shah N, Chang W, White V. et al. Orbital lipoma: 2 cases and review of literature. Ophthalmic Plast Reconstr Surg 2007; 23: 202-205
- 431 Franchi G, Sleilati E, Soupre V. et al. Melanotic neuroectodermal tumour of infancy involving the orbit and maxilla: surgical management and follow-up strategy. Br J Plast Surg 2002; 55: 526-529
- 432 Koob M, Fayard C, Pariente D. et al. Prenatal diagnosis of orbital melanotic neuroectodermal tumor in infancy. Ultrasound Obstet Gynecol 2015; 46: 247-252
- 433 Soles B, Wilson A, Lucas D. et al. Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 2018; 142: 1358-1363
- 434 NIH. Childhood Rhabdomyosarcoma Treatment (PDQ®)–Health Professional Version. In: National Cancer Institute at the National Institutes of Health; 2020
- 435 Al-Bargi H, Al-Maiman M. Orbital fibrosarcoma: a case report and literature review. Egypt J Oral Maxillofac Surg 2012; 3: 45-48
- 436 Bahrami A, Folpe A. Adult-type fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol 2010; 34: 1504-1513
- 437 Scruggs B, Ho S, Valenzuela A. Diagnostic challenges in primary orbital fibrosarcoma: a case report. Clin Ophthalmol 2014; 8: 2319-2323
- 438 Almater A, Alfaleh A, Alshomar K et al. Retinoblastoma: Update on Current Management, Retinoblastoma - Past, Present and Future. In Alkatan H, Hrsg.Retinoblastoma: IntechOpen; 2019
- 439 Cassoux N, Lumbroso L, Levy-Gabriel C. et al. Retinoblastoma: Update on Current Management. Asia-Pac. J Ophthalmol 2017; 6: 290-295
- 440 WHO. RETINOBLASTOMA - 2014 Review of Cancer Medicines on the WHO List of Essential Medicines. In: Union for International Cancer Control; 2014
- 441 Caignard A, Faguer R, Mercier P. et al. Optic nerve and visual pathways primary glioblastoma treated with radiotherapy and temozolomide chemotherapy. Eur J Ophthalmol 2014; 24: 637-640
- 442 Waheed S, Huang B, Zamora C. Glioblastoma involving the optic nerve. Am J Neuroradiol 2018; 41: e
- 443 Alireza M, Amelot A, Chauvet D. Poor prognosis and challenging treatment of optic nerve malignant gliomas: Literature review and case report series. World Neurosurg 2017; 97: e1-e6
- 444 Ashour O, Stalling M, Ramsey J. et al. Intraocular Medulloepithelioma. RadioGraphics 2018; 38: 194-199
- 445 Verdijk R. On the Classification and Grading of Medulloepithelioma of the Eye. Ocul Oncol Pathol 2016; 2: 190-193
- 446 Hei Y, Kang L, Yang X. Orbital alveolar soft part sarcoma: A report of 8 cases and review of the literature. Oncol Lett 2018; 15: 304-314
- 447 Inci E, Korkut N, Erem M. et al. [Alveolar soft tissue sarcoma]. HNO 2004; 52: 145-149
- 448 Rose A, Kabiru J, Rose G. Alveolar soft-part sarcoma of the orbit. Afr J Paediatr Surg 2011; 8: 82-84
- 449 Dirksen U, Jürgens H. S1-Leitlinie: Ewing-Sarkome des Kindes- und Jugendalters. Stand 06/2014, DOI: AWMF-Register Nr. 025/006
- 450 Kaliki S, Rathi S, Palkonda V. Primary orbital Ewing sarcoma family of tumors: a study of 12 cases. Eye 2018; 32: 615-621
- 451 Chaugule S, Putambekar A, Gavade S. et al. Primary Orbital Leiomyosarcoma in an Adult Male. Ophthal Plast Reconstr Surg 2019; 35: e27-e29
- 452 Klippenstein K, Wesley R, Glick A. Orbital Leiomyosarcoma After Retinoblastoma. Ophthalmic Surg Lasers Imaging. Retina 1999; 30: 579-583
- 453 Yeniad B, Tuncer S, Peksayar G. et al. Primary Orbital Leiomyosarcoma. Ophth. Plast Reconstr Surg 2009; 25: 154-155
- 454 Cai Y, McMenamin E, Geoff R. et al. Primary liposarcoma of the orbit: A clinicopathologic study of seven cases. Ann Diagn Pathol 2001; 5: 255-266
- 455 Garrity J, Henderson J, Cameron J. Henderson's Tumors of Primitive Mesoderm, Striated muscle, Adipose Tissue and Smooth muscle.In Garrity JA HJ, Cameron JD , Hrsg. Henderson's orbital tumors. 4th edition. AuflPhiladelphia: Lippincott Williams and Wilkins; 2007: 123–141
- 456 Alkatan H, Chaudhry I, Al-Qahtani A. Epithelioid sarcoma of the orbit. Ann Saudi Med 2011; 31: 187-189
- 457 Inazawa N, Hatakeyama N, Hori T. et al. Primary orbital neuroblastoma in a 1-month-old boy. Pediatr Int 2014; 56: 122-125
- 458 Vallinayagam M, Rao V, Pandian D. et al. Primary orbital neuroblastoma with intraocular extension. Indian J Ophthalmol 2015; 63: 684-686
- 459 Zhang N, Lin L. Presumed primary orbital neuroblastoma in a 20-month-old female. Ophthal Plast Reconstr Surg 2010; 26: 383-385
- 460 Rose A, Luthert P, Jayasena C. et al. Primary Orbital Melanoma: Presentation, Treatment, and Long-term Outcomes for 13 Patients. Front Oncol 2017; 7: 316
- 461 Kiratli H, Balci K, Güler G. Primary orbital endodermal sinus tumor (yolk sac tumor). J AAPOS 2008; 12: 623-625
- 462 Mogaddam A, Memar B, Aledavood A. et al. Isolated Orbital Endodermal Sinus Tumor. Ophthal Plast Reconstr Surg 2007; 23: 477-479
- 463 Eckardt A, Lemound J, Rana M. et al. Orbital lymphoma: diagnostic approach and treatment outcome. World J Surg Oncol. 2013 DOI: 73
- 464 Olsen T, Heegaard S. Orbitla Lymphoma. Surv Ophthalmol 2019; 64: 45-66
- 465 Sharma T, Kamath M. Diagnosis and Management of Orbital Lymphoma. Ophthalmic Pearls. 2015 DOI: 37-39
- 466 Bidar M, Wilson M, Laquis S. et al. Clinical and imaging characteristics of orbital leukemic tumors. Ophthalmic Plast Reconstr Surg 2007; 23: 87-93
- 467 Hatton M, Rubin P. Chronic Lymphocytic Leukemia of the Orbit. Arch Ophthalmol 2002; 120: 990-991
- 468 Maka E, Lukáts O, Tóth J. et al. Orbital tumour as initial manifestation of acute myeloid leukemia: granulocytic sarcoma: case report. Pathol Oncol Res 2008; 14: 209-211
- 469 Taylor C, Taylor R, Kinsey S. LEUKEMIC INFILTRATION OF THE ORBIT: Report of Three Cases and Literature Review. Pediatr Hematol Oncol 2005; 22: 415-422
- 470 Pausch N, Sterker I, Bauer U. Maligner intraorbitaler Tumor: Primum oder Metastase?. HNO 2016; 64: 262-264
- 471 Schenke T, Lehmann G, Kunze C. et al. Eine abszessverdächtige Raumforderung in der extraokulären Augenmuskulatur. HNO. 2020
- 472 Suarez C, Ferlito A, Lund V. Management of the orbit in malignant sinunasal tumors. Head Neck 2008; 30: 242-250
- 473 Evert K, Kühnel T, Weiß K. et al. Diagnostik und Management von Gefäßanomalien. Der. Pathologe 2019; 40: 422-430
- 474 Müller-Wille R, Wildgruber M, Sadick M. et al. Vascular Anomalies (Part II): Interventional Therapy of Peripheral Vascular Malformations. Fortschr Röntgenstr 2018; 190: 927-937
- 475 Sadick M, Müller-Wille R, Wildgruber M. et al. Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies. Fortschr Röntgenstr 2018; 190: 825-835
- 476 Wassef M, Blei F, Adams D. et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 2015; 136: e203-e214
- 477 Fay A, Nguyen J, Jakobiec F. et al. Propranolol for Isolated Orbital Infantile Hemangioma. ARCH OPHTHALMOL 2010; 128: 256-258
- 478 Grantzow R, Schmittenbecher P, Höger P. et al. S2k-Leitlinie 006/100: Infantile Hämangiome im Säuglings- und Kleinkindesalter. AWMF online. 2015 DOI: 1-13
- 479 Goldsmith J, van de Rijn M, Syed N. Orbital Hemangiopericytoma and Solitary Fibrous Tumor: A Morphologic Continuum. Internat J Surg Pathol 2001; 9: 295-302
- 480 Pacheco L, Fernandes B, Miyamoto C. et al. Rapid growth of an orbital hemangiopericytoma with atypical histopathological findings. Clin Ophthalmol 2014; 8: 31-33
- 481 Westerfeld C, Allen C, Mihm M. et al. Non-Involuting Congenital Hemangioma Presenting as an Orbital Mass in a Newborn. Investigative Ophthalmology & Visual Science 48: 3582
- 482 Gbolahan O, Fasina O, Adisa A. et al. Spindle cell hemangioma: Unusual presentation of an uncommon tumor. J Oral Maxillofac Pathol 2015; 19: 406
- 483 Budimir I, Demirović A, Iveković R. et al. Epithelioid hemangioma of the orbit: case report. Acta Clin Croat 2015; 54: 92-95
- 484 McEachren T, Brownstein S, Jordan D. et al. Epithelioid hemangioma of the orbit. Ophtalmology 2000; 107: 806-810
- 485 Stiefel H, Ng J, Wilson D. et al. Orbital Cellular Epithelioid Hemangioma. Ocul Oncol Pathol 2019; 5: 424-431
- 486 Kamano H, Noguchi T, Yoshiura T. et al. Intraorbital lobular capillary hemangioma (pyogenic granuloma). Radiat Med 2008; 26: 609-612
- 487 Häußler S, Uecker F, Knopke S. et al. Tufted angiomas of the head and neck. HNO 2018; 66: S1-S6
- 488 Benesch M, Lackner H, Sorantin E. et al. Medikamentöse Therapie vaskulärer Anomalien. Paediatr Paedolog 2020; 55: 21-27
- 489 Cho W, Kim S, Park S. et al. Intracranial kaposiform hemangioendothelioma: proposal of a new malignant variant. J Pediatr Ophthalmol Strabismus 2009; 3: 147-150
- 490 Li B, Li Y, Tian X. et al. Unusual multifocal intraosseous papillary intralymphatic angioendothelioma (Dabska tumor) of facial bones: a case report and review of literature. Diagn Pathol 2013; 8: 160
- 491 Collaco L, Goncalves M, Gomes L. Orbital Kaposi’s Sarcoma in Acquired Immunodeficiency Syndrome. Eur J Ophthalmol 2000; 10: 88-90
- 492 Siddens J, Fishman J, Jackson I. et al. Primary orbital angiosarcoma: a case report. Ophthalmic Plast Reconstr Surg 1999; 15: 454-459
- 493 Lyon D, Tang T, Kidder T. Epithelioid Hemangioendothelioma of the Orbital Bones. Ophtalmology 1992; 99: 1773-1778
- 494 ISSVA. ISSVA Classification of Vascular Anomalies ©2018. In: International Society for the Study of Vascular Anomalies
- 495 Wildgruber M, Sadick M, Muller-Wille R. et al. Vascular tumors in infants and adolescents. Insights Imaging 2019; 10: 30
- 496 Gilbert P, Dubois J, Giroux F. New Treatment Approaches to Arteriovenous Malformations. Semin Intervent Radiol 2017; 34: 258-271
- 497 Schobinger RA, Fontaine R. Periphere Angiodysplasien.
Korrespondenzadresse
Publication History
Article published online:
30 April 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
Literatur
- 1 Poloschek CM, Lagreze WA, Ridder GJ. et al. [Clinical and neuroradiological diagnostics of orbital tumors]. Ophthalmologe 2011; 108: 510-518
- 2 Plontke SK, Glien A, Kisser U. et al. [Diseases and Surgery of the Orbit]. Laryngorhinootologie 2020; 99: 896-917
- 3 AWMF. Rekonstruktion von Orbitadefekten (S2e-Leitlinie), Registernummer: 007/099. In: Arbeitsgemeinschaft medizinisch wissenschaftlicher Fachgesellschaften (AWMF); 2013
- 4 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 57–77
- 5 Bonavolonta G, Tranfa F, de Conciliis C. et al. Dermoid cysts: 16-year survey. Ophthalmic Plast Reconstr Surg 1995; 11: 187-192
- 6 Jordan DR, Spitellie P, Brownstein S. et al. Orbital cholesterol granuloma and cholesteatoma: significance of differentiating the two. Ophthalmic Plast Reconstr Surg 2007; 23: 415-417
- 7 Sherman RP, Rootman J, Lapointe JS. Orbital dermoids: clinical presentation and management. Br J Ophthalmol 1984; 68: 642-652
- 8 Shields JA, Kaden IH, Eagle RC. et al. Orbital dermoid cysts: clinicopathologic correlations, classification, and management. The 1997 Josephine E. Schueler Lecture. Ophthalmic Plast Reconstr Surg 1997; 13: 265-276
- 9 Delfini R, Missori P, Iannetti G. et al. Mucoceles of the paranasal sinuses with intracranial and intraorbital extension: report of 28 cases. Neurosurgery 1993; 32: 901-906 discussion 906
- 10 Heichel J, Struck HG, Hammer T. et al. [Pediatric acute dacryocystitis due to frontoethmoidal mucocele]. HNO 2019; 67: 458-462
- 11 Nicollas R, Facon F, Sudre-Levillain I. et al. Pediatric paranasal sinus mucoceles: etiologic factors, management and outcome. Int J Pediatr Otorhinolaryngol 2006; 70: 905-908
- 12 Holtmann C, Brachert M, Spaniol K. et al. [Dacryolith in a lacrimal duct]. Ophthalmologe 2016; 113: 244-247
- 13 Tanaboonyawat S, Idowu OO, Copperman TS. et al. Dacryops - A review. Orbit 2020; 39: 128-134
- 14 Junemann A, Holbach LM. [Epithelial giant inclusion cyst 50 years after enucleation without orbital implant]. Klin Monbl Augenheilkd 1998; 212: 127-128
- 15 Kalantzis GK, Verity DH, Rose GE. Periocular implantation cysts: a late complication of ophthalmic surgery. Eye (Lond) 2014; 28: 1004-1007
- 16 Gupta R, Seith A, Guglani B. et al. Congenital cystic eye: features on MRI. Br J Radiol 2007; 80: e137-140
- 17 Guthoff R, Klein R, Lieb WE. Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 2004; 242: 268-271
- 18 Kuchle HJ, Normann J, Lubbering I. [Congenital cystic eye]. Klin Monbl Augenheilkd 1986; 188: 239-241
- 19 Schittkowski MP, Guthoff RF. Injectable self inflating hydrogel pellet expanders for the treatment of orbital volume deficiency in congenital microphthalmos: preliminary results with a new therapeutic approach. Br J Ophthalmol 2006; 90: 1173-1177
- 20 Schittkowski MP, Guthoff RF. Systemic and ophthalmological anomalies in congenital anophthalmic or microphthalmic patients. Br J Ophthalmol 2010; 94: 487-493
- 21 Badtke G, Tost M. Anophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 328–344
- 22 Badtke G, Tost M. Mikrophthalmus congenitus.In Velhagen K , Hrsg. Der Augenarzt, Band XI. Leipzig: VEB Georg Thieme; 1986: 312–328
- 23 Chaudhry IA, Arat YO, Shamsi FA. et al. Congenital microphthalmos with orbital cysts: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2004; 20: 452-457
- 24 Oguz H, Ozturk A, San I. Congenital nasolacrimal duct occlusion with clinical anophthalmos: a possible new association. Ophthalmic Genet 2003; 24: 181-185
- 25 Pejic M, Luecke K, Meoded A. et al. Pediatric Cephaloceles: A Multimodality Review. Appl Radiol 2020; 49: 26-32
- 26 Terry A, Patrinely JR, Anderson RL. et al. Orbital meningoencephalocele manifesting as a conjunctival mass. Am J Ophthalmol 1993; 115: 46-49
- 27 Vashisht S, Ghai S, Hatimota P. et al. Cystic lesions of the orbit : A CT spectrum. Indian Journal of Radiology and Imaging 2003; 13: 139
- 28 Modi P, Shah NA, Bhalodia JN. et al. ORBITAL TUMORS IN CHILDREN: A DESCRIPTIVE STUDY AT TERTIARY CARE CENTRE. The Journal of medical research 2013; 3: 362-366
- 29 Green WR, Zimmerman LE. Ectopic lacrimal gland tissue. Report of eight cases with orbital involvement. Arch Ophthalmol 1967; 78: 318-327
- 30 Scheiner AJ, Frayer WC, Rorke LB et al. Ectopic brain tissue in the orbit. Eye (Lond) 1999; 13: ( Pt 2) 251–254
- 31 Damato PJ, Damato FJ. Neonatal Orbital Teratoma. Br J Ophthalmol 1962; 46: 685-691
- 32 Herman TE, Vachharajani A, Siegel MJ. Massive congenital orbital teratoma. J Perinatol 2009; 29: 396-397
- 33 Sesenna E, Ferri A, Thai E. et al. Huge orbital teratoma with intracranial extension: a case report. J Pediatr Surg 2010; 45: e27-31
- 34 Duke-Elder S. Anomalies of the Bony Orbit. In: Duke-Elder S, Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 942-949
- 35 Exner S, Bogusch G, Sokiranski R. Cribra orbitalia visualized in computed tomography. Ann Anat 2004; 186: 169-172
- 36 Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol 2004; 88: 833-834
- 37 Duke-Elder S. Deformities of the Face.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964; 1007–1035
- 38 Kawamoto HK. The kaleidoscopic world of rare craniofacial clefts: order out of chaos (Tessier classification). Clin Plast Surg 1976; 3: 529-572
- 39 Fearon JA. Rare craniofacial clefts: a surgical classification. J Craniofac Surg 2008; 19: 110-112
- 40 Tessier P. Anatomical classification facial, cranio-facial and latero-facial clefts. J Maxillofac Surg 1976; 4: 69-92
- 41 Heike CL, Hing AV, Aspinall CA. et al. Clinical care in craniofacial microsomia: a review of current management recommendations and opportunities to advance research. Am J Med Genet C Semin Med Genet 2013; 163C: 271-282
- 42 Dupont S, Catala M, Hasboun D. et al. Progressive facial hemiatrophy and epilepsy: a common underlying dysgenetic mechanism. Neurology 1997; 48: 1013-1018
- 43 Fea AM, Aragno V, Briamonte C. et al. Parry Romberg syndrome with a wide range of ocular manifestations: a case report. BMC Ophthalmol 2015; 15: 119
- 44 Duke-Elder S. Deformities of the Skull.In Duke-Elder S , Hrsg. System of Ophthalmology Vol III: Normal and abnormal Development Part 2, Congenital Deformities: Henry Kimpton. 1964: 1035–1072
- 45 Freudlsperger C, Castrillón-Oberndorfer G, Hoffmann J. et al. Isolierte, nichtsyndromale Kraniosynostosen. Der MKG-Chirurg 2013; 6: 301-313
- 46 Governale LS. Craniosynostosis. Pediatr Neurol 2015; 53: 394-401
- 47 Kotrikova B, Muhling J. [Surgery of craniofacial deformities]. Klin Monbl Augenheilkd 2004; 221: 970-977
- 48 Wilkie AOM, Johnson D, Wall SA. Clinical genetics of craniosynostosis. Curr Opin Pediatr 2017; 29: 622-628
- 49 Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999; 36: 177-182
- 50 Dubourg C, Bendavid C, Pasquier L. et al. Holoprosencephaly. Orphanet J Rare Dis 2007; 2: 8
- 51 Lustig LR, Holliday MJ, McCarthy EF. et al. Fibrous dysplasia involving the skull base and temporal bone. Arch Otolaryngol Head Neck Surg 2001; 127: 1239-1247
- 52 Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis 2009; 4: 5
- 53 Lannon DA, Earley MJ. Cherubism and its charlatans. Br J Plast Surg 2001; 54: 708-711
- 54 Grass S, Welkoborsky HJ, Mobius H. et al. [Orbital complications]. HNO 2018; 66: 800-811
- 55 Lehnerdt G, Peraud A, Berghaus A. et al. [Orbital and intracranial complications of acute sinusitis. Diagnostics and therapy in children and adolescents]. HNO 2011; 59: 75-86 quiz 87-78
- 56 Stammberger H. [Complications of inflammatory paranasal sinus diseases including iatrogenically-induced complications]. Eur Arch Otorhinolaryngol Suppl 1993; 1: 61-102
- 57 Welkoborsky HJ, Grass S, Deichmuller C. et al. Orbital complications in children: differential diagnosis of a challenging disease. Eur Arch Otorhinolaryngol 2015; 272: 1157-1163
- 58 Hamed-Azzam S, AlHashash I, Briscoe D. et al. Rare Orbital Infections ~ State of the Art ~ Part II. J Ophthalmic Vis Res 2018; 13: 183-190
- 59 Rootman J. Diseases of the orbit.Second edition Aufl. Lippincott Williams & Wilkins; Philadelphia: 2003
- 60 Madge SN, Prabhakaran VC, Shome D. et al. Orbital tuberculosis: a review of the literature. Orbit 2008; 27: 267-277
- 61 Fay A, Dolman PJ. Diseases and Disorders of the Orbit and Ocular Adnexa Expert Consult. In Fay A, Dolman PJ (eds). Elsevier; 2017
- 62 Heichel J, Griebsch M, Siebolts U. et al. [Bilateral periocular necrotizing fasciitis with unilateral orbital involvement]. Ophthalmologe 2020; 117: 1121-1125
- 63 Huber L, Budjan J, Rotter N. et al. [Necrotizing fasciitis in the head and neck region-three case reports and a review of the literature]. HNO 2020; 68: 935-943
- 64 Amrith S, Hosdurga Pai V, Ling WW. Periorbital necrotizing fasciitis -- a review. Acta Ophthalmol 2013; 91: 596-603
- 65 Wong CH, Chang HC, Pasupathy S. et al. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85: 1454-1460
- 66 Lazzeri D, Lazzeri S, Figus M. et al. Periorbital necrotising fasciitis. Br J Ophthalmol 2010; 94: 1577-1585
- 67 Wladis EJ, Levin F, Shinder R. Clinical Parameters and Outcomes in Periorbital Necrotizing Fasciitis. Ophthalmic Plast Reconstr Surg 2015; 31: 467-469
- 68 Federspil P. German Society of Oto-Rhino-Laryngology H, Neck S. [Guidelines: Antibiotic treatment of infections of the head and neck: consensus report on behalf of the presidency of the German Society of Oto-Rhino-Laryngology, Head and Neck Surgery edited by P. Federspil, Homburg/Saar]. . HNO 2009; 57: 377-394
- 69 Barrani M, Massei F, Scaglione M. et al. Unusual onset of a case of chronic recurrent multifocal osteomyelitis. Pediatr Rheumatol Online J 2015; 13: 60
- 70 Dagan O, Barak Y, Metzker A. Pyoderma gangrenosum and sterile multifocal osteomyelitis preceding the appearance of Takayasu arteritis. Pediatr Dermatol 1995; 12: 39-42
- 71 Huber AM, Lam PY, Duffy CM. et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr 2002; 141: 198-203
- 72 Hu AC, Ng WKY. Orbital Osteomyelitis in the Pediatric Patient. J Craniofac Surg 2021; 32: 206-209
- 73 Cornely OA, Alastruey-Izquierdo A, Arenz D. et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect Dis 2019; 19: e405-e421
- 74 Bakhshaee M, Bojdi A, Allahyari A. et al. Acute invasive fungal rhinosinusitis: our experience with 18 cases. Eur Arch Otorhinolaryngol 2016; 273: 4281-4287
- 75 van Burik JA, Hare RS, Solomon HF. et al. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis 2006; 42: e61-65
- 76 Songu M, Unlu HH, Gunhan K. et al. Orbital exenteration: A dilemma in mucormycosis presented with orbital apex syndrome. Am J Rhinol 2008; 22: 98-103
- 77 Nicolai P, Lombardi D, Tomenzoli D. et al. Fungus ball of the paranasal sinuses: experience in 160 patients treated with endoscopic surgery. Laryngoscope 2009; 119: 2275-2279
- 78 Johnson TE, Casiano RR, Kronish JW. et al. Sino-orbital aspergillosis in acquired immunodeficiency syndrome. Arch Ophthalmol 1999; 117: 57-64
- 79 Levin LA, Avery R, Shore JW. et al. The spectrum of orbital aspergillosis: a clinicopathological review. Surv Ophthalmol 1996; 41: 142-154
- 80 Siddiqui AA, Shah AA, Bashir SH. Craniocerebral aspergillosis of sinonasal origin in immunocompetent patients: clinical spectrum and outcome in 25 cases. Neurosurgery 2004; 55: 602-611 discussion 611-603
- 81 Nielsen EW, Weisman RA, Savino PJ. et al. Aspergillosis of the sphenoid sinus presenting as orbital pseudotumor. Otolaryngol Head Neck Surg 1983; 91: 699-703
- 82 Sivak-Callcott JA, Livesley N, Nugent RA. et al. Localised invasive sino-orbital aspergillosis: characteristic features. Br J Ophthalmol 2004; 88: 681-687
- 83 Lamoth F, Calandra T. Early diagnosis of invasive mould infections and disease. J Antimicrob Chemother 2017; 72: i19-i28
- 84 McCarthy M, Rosengart A, Schuetz AN. et al. Mold infections of the central nervous system. N Engl J Med 2014; 371: 150-160
- 85 Green WR, Font RL, Zimmerman LE. Asperillosis of the orbit. Report of ten cases and review of the literature. Arch Ophthalmol 1969; 82: 302-313
- 86 Denning DW. Therapeutic outcome in invasive aspergillosis. Clin Infect Dis 1996; 23: 608-615
- 87 Robert Koch-Institut. Infektionsepidemiologisches Jahrbuch meldepflichtiger Krankheiten für 2019 Berlin: Robert Koch-Institut; 2020
- 88 Gomez Morales A, Croxatto JO, Crovetto L. et al. Hydatid cysts of the orbit. A review of 35 cases. Ophthalmology 1988; 95: 1027-1032
- 89 Xiao A, Xueyi C. Hydatid cysts of the orbit in Xinjiang: a review of 18 cases. Orbit 1999; 18: 151-155
- 90 Kars Z, Kansu T, Ozcan OE. et al. Orbital echinococcosis. Report of two cases studied by computerized tomography. J Clin Neuroophthalmol 1982; 2: 197-199
- 91 Chtira K, Benantar L, Aitlhaj H. et al. The surgery of intra-orbital hydatid cyst: a case report and literature review. Pan Afr Med J 2019; 33: 167
- 92 Senyuz OF, Yesildag E, Celayir S. Albendazole therapy in the treatment of hydatid liver disease. Surg Today 2001; 31: 487-491
- 93 Pushker N, Bajaj MS, Balasubramanya R. Disseminated cysticercosis involving orbit, brain and subcutaneous tissue. J Infect 2005; 51: e245-248
- 94 Walrath JD, Lalin SC, Leib ML. Cysticercosis isolated to the orbit. Ophthalmic Plast Reconstr Surg 2003; 19: 243-244
- 95 Sotelo J, Escobedo F, Rodriguez-Carbajal J. et al. Therapy of parenchymal brain cysticercosis with praziquantel. N Engl J Med 1984; 310: 1001-1007
- 96 Nentwich MM, Pleyer U, Schaller UC. et al. [International ophthalmology and travel medicine]. Ophthalmologe 2016; 113: 83-94
- 97 Mombaerts I, Rose GE, Garrity JA. Orbital inflammation: Biopsy first. Surv Ophthalmol 2016; 61: 664-669
- 98 Ben Simon GJ, Yoon MK, Atul J. et al. Clinical manifestations of orbital mass lesions at the Jules Stein Eye Institute, 1999-2003. Ophthalmic Surg Lasers Imaging 2006; 37: 25-32
- 99 Nölle B, Both M. Idiopathische Orbitaentzündung.In Pleyer U , Hrsg. Entzündliche Augenerkrankungen. Berlin, Heidelberg: Springer- Verlag; 2016: 48–57
- 100 Swamy BN, McCluskey P, Nemet A. et al. Idiopathic orbital inflammatory syndrome: clinical features and treatment outcomes. Br J Ophthalmol 2007; 91: 1667-1670
- 101 Fujii H, Fujisada H, Kondo T. et al. Orbital pseudotumor: histopathological classification and treatment. Ophthalmologica 1985; 190: 230-242
- 102 Mombaerts I, Goldschmeding R, Schlingemann RO. et al. What is orbital pseudotumor?. Surv Ophthalmol 1996; 41: 66-78
- 103 Rootman J, Nugent R. The classification and management of acute orbital pseudotumors. Ophthalmology 1982; 89: 1040-1048
- 104 Rootman J, McCarthy M, White V. et al. Idiopathic sclerosing inflammation of the orbit. A distinct clinicopathologic entity. Ophthalmology 1994; 101: 570-584
- 105 Iaconetta G, Stella L, Esposito M. et al. Tolosa-Hunt syndrome extending in the cerebello-pontine angle. Cephalalgia 2005; 25: 746-750
- 106 Speth MM, Metternich FU, Mina R. et al. Tolosa-Hunt-Syndrom, das sich als migrierende, auf Kortikosteroid reagierende CN3- und CN6-Lähmung bei verschatteten Keilbeinhöhlen darstellt. forum hno: omnimed Verlag 05/2020;
- 107 Halabi T, Sawaya R. Successful Treatment of Tolosa-Hunt Syndrome after a Single Infusion of Infliximab. J Clin Neurol 2018; 14: 126-127
- 108 Mormont E, Laloux P, Vauthier J. et al. Radiotherapy in a case of Tolosa-Hunt syndrome. Cephalalgia 2000; 20: 931-933
- 109 Perez CA, Evangelista M. Evaluation and Management of Tolosa-Hunt Syndrome in Children: A Clinical Update. Pediatr Neurol 2016; 62: 18-26
- 110 Zhang X, Zhang W, Liu R. et al. Factors that influence Tolosa-Hunt syndrome and the short-term response to steroid pulse treatment. J Neurol Sci 2014; 341: 13-16
- 111 Espinoza GM, Liu JL. Orbital Vasculitides-Differential Diagnosis. Curr Rheumatol Rep 2019; 21: 54
- 112 Tiegs-Heiden CA, Eckel LJ, Hunt CH. et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014; 35: 1393-1397
- 113 Erdei A, Steiber Z, Molnar C. et al. Exophthalmos in a young woman with no graves' disease - a case report of IgG4-related orbitopathy. BMC Ophthalmol 2018; 18: 5
- 114 Jurkov M, Olze H, Klauschen F. et al. [IgG4-Related Orbitopathy as an Important Differential Diagnosis of Advanced Silent Sinus Syndrome. German version]. HNO 2020; 68: 864-868
- 115 Mulholland GB, Jeffery CC, Satija P. et al. Immunoglobulin G4-related diseases in the head and neck: a systematic review. J Otolaryngol Head Neck Surg 2015; 44: 24
- 116 Detiger SE, Karim AF, Verdijk RM. et al. The treatment outcomes in IgG4-related orbital disease: a systematic review of the literature. Acta Ophthalmol 2019; 97: 451-459
- 117 Plaza JA, Garrity JA, Dogan A. et al. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol 2011; 129: 421-428
- 118 Perumal B, Black EH, Levin F. et al. Non-infectious orbital vasculitides. Eye (Lond) 2012; 26: 630-639
- 119 Sfiniadaki E, Tsiara I, Theodossiadis P. et al. Ocular Manifestations of Granulomatosis with Polyangiitis: A Review of the Literature. Ophthalmol Ther 2019; 8: 227-234
- 120 Mukhtyar C, Flossmann O, Hellmich B. et al. Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: a systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67: 1004-1010
- 121 Holle JU, Voigt C, Both M. et al. Orbital masses in granulomatosis with polyangiitis are associated with a refractory course and a high burden of local damage. Rheumatology (Oxford) 2013; 52: 875-882
- 122 Perry SR, Rootman J, White VA. The clinical and pathologic constellation of Wegener granulomatosis of the orbit. Ophthalmology 1997; 104: 683-694
- 123 Chang SY, Keogh KA, Lewis JE. et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener's): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol 2013; 44: 2432-2437
- 124 Martinez Del Pero M, Chaudhry A, Jones RB. et al. B-cell depletion with rituximab for refractory head and neck Wegener's granulomatosis: a cohort study. Clin Otolaryngol 2009; 34: 328-335
- 125 Takanashi T, Uchida S, Arita M. et al. Orbital inflammatory pseudotumor and ischemic vasculitis in Churg-Strauss syndrome: report of two cases and review of the literature. Ophthalmology 2001; 108: 1129-1133
- 126 Cottin V, Cordier JF. Churg-Strauss syndrome. Allergy 1999; 54: 535-551
- 127 Masi AT, Hunder GG, Lie JT. et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094-1100
- 128 Vaglio A, Moosig F, Zwerina J. Churg-Strauss syndrome: update on pathophysiology and treatment. Curr Opin Rheumatol 2012; 24: 24-30
- 129 Guillevin L, Lhote F, Gayraud M. et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17-28
- 130 Nagel MA, White T, Khmeleva N. et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol 2015; 72: 1281-1287
- 131 Borruat FX, Bogousslavsky J, Uffer S. et al. Orbital infarction syndrome. Ophthalmology 1993; 100: 562-568
- 132 Stone JH, Tuckwell K, Dimonaco S. et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med 2017; 377: 317-328
- 133 Seyahi E. Takayasu arteritis: an update. Curr Opin Rheumatol 2017; 29: 51-56
- 134 Tugal-Tutkun I. Systemic vasculitis and the eye. Curr Opin Rheumatol 2017; 29: 24-32
- 135 Misra DP, Wakhlu A, Agarwal V. et al. Recent advances in the management of Takayasu arteritis. Int J Rheum Dis 2019; 22 (Suppl. 01) 60-68
- 136 Iliescu DA, Timaru CM, Batras M. et al. Cogan's Syndrome. Rom J Ophthalmol 2015; 59: 6-13
- 137 Tayer-Shifman OE, Ilan O, Tovi H. et al. Cogan's syndrome--clinical guidelines and novel therapeutic approaches. Clin Rev Allergy Immunol 2014; 47: 65-72
- 138 Grasland A, Pouchot J, Hachulla E. et al. Typical and atypical Cogan's syndrome: 32 cases and review of the literature. Rheumatology (Oxford) 2004; 43: 1007-1015
- 139 Gluth MB, Baratz KH, Matteson EL. et al. Cogan syndrome: a retrospective review of 60 patients throughout a half century. Mayo Clin Proc 2006; 81: 483-488
- 140 Padoan R, Cazzador D, Pendolino AL. et al. Cogan's syndrome: new therapeutic approaches in the biological era. Expert Opin Biol Ther 2019; 19: 781-788
- 141 Kokturk A. Clinical and Pathological Manifestations with Differential Diagnosis in Behcet's Disease. Patholog Res Int 2012; 2012: 690390
- 142 Hammami S, Yahia SB, Mahjoub S. et al. Orbital inflammation associated with Behcet's disease. Clin Exp Ophthalmol 2006; 34: 188-190
- 143 Urruticoechea-Arana A, Cobo-Ibanez T, Villaverde-Garcia V. et al. Efficacy and safety of biological therapy compared to synthetic immunomodulatory drugs or placebo in the treatment of Behcet's disease associated uveitis: a systematic review. Rheumatol Int 2019; 39: 47-58
- 144 Bettiol A, Silvestri E, Di Scala G. et al. The right place of interleukin-1 inhibitors in the treatment of Behcet's syndrome: a systematic review. Rheumatol Int 2019; 39: 971-990
- 145 Hedrich CM, Schnabel A, Hospach T. Kawasaki Disease. Front Pediatr 2018; 6: 198
- 146 Demirsoy S, Gucuyener K, Olgunturk R. et al. A case of Kawasaki syndrome associated with preseptal cellulitis in orbita. Turk J Pediatr 1988; 30: 55-59
- 147 Felz MW, Patni A, Brooks SE. et al. Periorbital vasculitis complicating Kawasaki syndrome in an infant. Pediatrics 1998; 101: E9
- 148 Sheard RM, Pandey KR, Barnes ND. et al. Kawasaki disease presenting as orbital cellulitis. J Pediatr Ophthalmol Strabismus 2000; 37: 123-125
- 149 Son MB, Gauvreau K, Burns JC. et al. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J Pediatr 2011; 158: 644-649 e641
- 150 Read RW. Clinical mini-review: systemic lupus erythematosus and the eye. Ocul Immunol Inflamm 2004; 12: 87-99
- 151 Arthurs BP, Khalil MK, Chagnon F. et al. Orbital infarction and melting in a patient with systemic lupus erythematosus. Ophthalmology 1999; 106: 2387-2390
- 152 Nabili S, McCarey DW, Browne B. et al. A case of orbital myositis associated with rheumatoid arthritis. Ann Rheum Dis 2002; 61: 938-939
- 153 Sanchez IJ, Finger DR, Bradshaw DJ. Orbital inflammatory syndrome and systemic lupus erythematosus. J Clin Rheumatol 1995; 1: 295-298
- 154 Kokotis P, Theodossiadis P, Bouros C. et al. Bilateral ocular myositis as a late complication of dermatomyositis. J Rheumatol 2005; 32: 379-381
- 155 Lokdarshi G, Pushker N, Bajaj MS. Sclerosing Lesions of the Orbit: A Review. Middle East Afr J Ophthalmol 2015; 22: 447-451
- 156 Petrarolha SM, Rodrigues BS, Filho FD. et al. Unilateral Eyelid Edema as Initial Sign of Orbital Sarcoidosis. Case Rep Ophthalmol Med 2016; 2016: 6912927
- 157 Rosenbaum JT, Choi D, Wilson DJ. et al. Parallel Gene Expression Changes in Sarcoidosis Involving the Lacrimal Gland, Orbital Tissue, or Blood. JAMA Ophthalmol 2015; 133: 770-777
- 158 Mavrikakis I, Rootman J. Diverse clinical presentations of orbital sarcoid. Am J Ophthalmol 2007; 144: 769-775
- 159 Demirci H, Christianson MD. Orbital and adnexal involvement in sarcoidosis: analysis of clinical features and systemic disease in 30 cases. Am J Ophthalmol 2011; 151: 1074-1080 e1071
- 160 McNab AA. The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond) 2018; 32: 248-261
- 161 Pasadhika S, Rosenbaum JT. Ocular Sarcoidosis. Clin Chest Med 2015; 36: 669-683
- 162 Aluclu MU, Keklikci U, Guzel A. et al. Melkersson-Rosenthal syndrome with partial oculomotor nerve palsy. Ann Saudi Med 2008; 28: 135-137
- 163 Shapiro M, Peters S, Spinelli HM. Melkersson-Rosenthal syndrome in the periocular area: a review of the literature and case report. Ann Plast Surg 2003; 50: 644-648
- 164 Cocuroccia B, Gubinelli E, Annessi G. et al. Persistent unilateral orbital and eyelid oedema as a manifestation of Melkersson-Rosenthal syndrome. J Eur Acad Dermatol Venereol 2005; 19: 107-111
- 165 Pierre-Filho Pde T, Rocha EM, Natalino R. et al. Upper eyelid oedema in Melkersson-Rosenthal syndrome. Clin Exp Ophthalmol 2004; 32: 439-440
- 166 Khandpur S, Malhotra AK, Khanna N. Melkersson-Rosenthal syndrome with diffuse facial swelling and multiple cranial nerve palsies. J Dermatol 2006; 33: 411-414
- 167 Rawlings NG, Valenzuela AA, Allen LH. et al. Isolated eyelid edema in Melkersson-Rosenthal syndrome: a case series. Eye (Lond) 2012; 26: 163-166
- 168 Ramaswamy B, Singh R, Manusrut M. et al. Sclerosing lipogranuloma of the eyelid: unusual complication following nasal packing in endoscopic sinus surgery. BMJ Case Rep. 2015 2015.
- 169 Witschel H, Geiger K. Paraffin induced sclerosing lipogranuloma of eyelids and anterior orbit following endonasal sinus surgery. Br J Ophthalmol 1994; 78: 61-65
- 170 Herrmann B, Rozot P, Baylac F. et al. [Lipogranuloma of the orbit. Apropos of a case]. J Fr Ophtalmol 1996; 19: 780-784
- 171 AlGhafri L, Galindo-Ferreiro A, Maktabi A. et al. Idiopathic orbital lipogranuloma. Int J Ophthalmol 2017; 10: 494-496
- 172 Jakobiec FA, Rai R, Rashid A. et al. Bilateral Eyelid Pseudoptosis From Lipogranulomas of the Preaponeurotic Fat Pads. Ophthalmic Plast Reconstr Surg 2015; 31: e125-131
- 173 Paik JS, Cho WK, Park GS. et al. Eyelid-associated complications after autogenous fat injection for cosmetic forehead augmentation. BMC Ophthalmol 2013; 13: 32
- 174 Park JY, Kim N. Periorbital Lipogranuloma after Facial Autologous Fat Injection and Its Treatment Outcomes. Korean J Ophthalmol 2016; 30: 10-16
- 175 Cafaro G, Croia C, Argyropoulou OD. et al. One year in review 2019: Sjogren's syndrome. Clin Exp Rheumatol 2019; 37 (Suppl. 118) 3-15
- 176 Mathews PM, Hahn S, Hessen M. et al. Ocular complications of primary Sjogren syndrome in men. Am J Ophthalmol 2015; 160: 447-452 e441
- 177 Danlos FX, Daoued F, Seror R. et al. Orbital Myositis and Primary Sjogren Syndrome. J Rheumatol 2015; 42: 1536-1537
- 178 Tonami H, Matoba M, Kuginuki Y. et al. Clinical and imaging findings of lymphoma in patients with Sjogren syndrome. J Comput Assist Tomogr 2003; 27: 517-524
- 179 Luemsamran P, Rootman J, White VA. et al. The role of biopsy in lacrimal gland inflammation: A clinicopathologic study. Orbit 2017; 36: 411-418
- 180 Ohshima K, Matsuo N, Yokoe S. et al. [A case of lacrimal gland malignant lymphoma, associated with Sjogren's syndrome]. Nippon Ganka Gakkai Zasshi 1991; 95: 386-392
- 181 Carubbi F, Cipriani P, Marrelli A. et al. Efficacy and safety of rituximab treatment in early primary Sjogren's syndrome: a prospective, multi-center, follow-up study. Arthritis Res Ther 2013; 15: R172
- 182 Kimura T. On the unusual granulations combined with hyperplastic changes of lymphatic tissue. Trans Soc Pathol Jpn 1948; 37: 179-180
- 183 Buder K, Ruppert S, Trautmann A. et al. Angiolymphoid hyperplasia with eosinophilia and Kimura's disease - a clinical and histopathological comparison. J Dtsch Dermatol Ges 2014; 12: 224-228
- 184 Claros P, Fokouo V, Nyada F. et al. Kimura's disease of the orbit: A modern diagnostic challenge. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 287-289
- 185 Googe PB, Harris NL, Mihm MC. Kimura's disease and angiolymphoid hyperplasia with eosinophilia: two distinct histopathological entities. J Cutan Pathol 1987; 14: 263-271
- 186 Kodama T, Kawamoto K. Kimura's disease of the lacrimal gland. Acta Ophthalmol Scand 1998; 76: 374-377
- 187 Monzen Y, Kiya K, Nishisaka T. Kimura's Disease of the Orbit Successfully Treated with Radiotherapy Alone: A Case Report. Case Rep Ophthalmol 2014; 5: 87-91
- 188 Bullock JD, Bartley GB, Campbell RJ. et al. Necrobiotic xanthogranuloma with paraproteinemia. Case report and a pathogenetic theory. Ophthalmology 1986; 93: 1233-1236
- 189 Shams PN, Rasmussen SL, Dolman PJ. Adult-Onset Asthma Associated With Simultaneous Conjunctival, Eyelid, and Orbital Xanthogranulomatosis Responsive to Systemic Immunosuppression. Ophthalmic Plast Reconstr Surg 2015; 31: e162-163
- 190 Elner VM, Mintz R, Demirci H. et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Ophthalmic Plast Reconstr Surg 2006; 22: 36-40
- 191 Efebera Y, Blanchard E, Allam C. et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk 2011; 11: 298-302
- 192 Cives M, Simone V, Rizzo FM. et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol 2015; 95: 1-11
- 193 Estrada-Veras JI, O'Brien KJ, Boyd LC. et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv 2017; 1: 357-366
- 194 Merritt H, Pfeiffer ML, Richani K. et al. Erdheim-Chester disease with orbital involvement: Case report and ophthalmic literature review. Orbit 2016; 35: 221-226
- 195 Manousaridis K, Casper J, Schittkowski MP. et al. [Erdheim-Chester disease of the orbit with compressive optic neuropathy]. Ophthalmologe 2010; 107: 266-269
- 196 Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D. et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75: 157-169
- 197 Altman J, Winkelmann RK. Diffuse normolipemic plane xanthoma. Generalized xanthelasma. Arch Dermatol 1962; 85: 633-640
- 198 Zak IT, Altinok D, Neilsen SS. et al. Xanthoma disseminatum of the central nervous system and cranium. AJNR Am J Neuroradiol 2006; 27: 919-921
- 199 Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol 2011; 147: 459-464
- 200 Minkov M, Lehrnbecher T. S1-Leitlinie 025/015 „Langerhanszell-Histiozytose (LCH) im Kindesalter In. 2017
- 201 Bertelmann E, Minko N, Lohneis P. et al. Benigne Neoplasien der Orbita. Klin Monatsbl Augenheilkd 2011; 228: 1113-1130
- 202 Maccheron LJ, McNab AA, Elder J. et al. Ocular adnexal Langerhans cell histiocytosis clinical features and management. Orbit 2006; 25: 169-177
- 203 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthalmic Plast Reconstr Surg 2016; 32: 28-34
- 204 Vemuganti GK, Naik MN, Honavar SG. Rosai dorfman disease of the orbit. J Hematol Oncol 2008; 1: 7
- 205 Bothra N, Kaliki S, Gowrishankar S. et al. Isolated unilateral eyelid Rosai-Dorfman disease. Oman J Ophthalmol 2018; 11: 300-302
- 206 Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63-70
- 207 Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19-73
- 208 McClellan SF, Ainbinder DJ. Orbital Rosai-Dorfman disease: a literature review. Orbit 2013; 32: 341-346
- 209 Ezeanosike E, Ezeanosike OB, Akpan SI. et al. Rhabdomyoblastic differentiation in rosai dorfman disease of the orbit in a 12-year-old male. Niger J Clin Pract 2018; 21: 1670-1673
- 210 Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123: 2924-2933
- 211 Goel R, Raut A, Agarwal A. et al. A Rare Presentation of Orbital Castleman's Disease. Case Rep Ophthalmol Med 2020; 2020: 1012759
- 212 Schrock A, Gutgemann I, Keiner S. [Castleman's disease in ear, nose, and throat practice]. HNO 2007; 55 (Suppl. 01) E29-32
- 213 Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman's disease: a systematic review of 404 published cases. Ann Surg 2012; 255: 677-684
- 214 van Rhee F, Wong RS, Munshi N. et al. Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014; 15: 966-974
- 215 Kesper C, Viestenz A, Siebolts U. et al. [Orbital Amyloidosis - Comparison of Two Different Clinical Courses]. Klin Monbl Augenheilkd 2020; 237: 35-40
- 216 Leibovitch I, Selva D, Goldberg RA. et al. Periocular and orbital amyloidosis: clinical characteristics, management, and outcome. Ophthalmology 2006; 113: 1657-1664
- 217 Mora-Horna ER, Rojas-Padilla R, Lopez VG. et al. Ocular adnexal and orbital amyloidosis: a case series and literature review. Int Ophthalmol 2016; 36: 281-298
- 218 Naxer S, Behnes CL, Schittkowski MP. [Amyloidosis--a rare differential diagnosis of an orbital tumour]. Klin Monbl Augenheilkd 2011; 228: 555-564
- 219 Ladner-Merz S, Muller-Ladner U. [Amyloidoses]. Z Rheumatol 2008; 67: 677-682 quiz 683
- 220 Khaira M, Mutamba A, Meligonis G. et al. The use of radiotherapy for the treatment of localized orbital amyloidosis. Orbit 2008; 27: 432-437
- 221 Eghbali A, Hassan S, Seehra G. et al. Ophthalmological findings in Gaucher disease. Mol Genet Metab 2019; 127: 23-27
- 222 Prakalapakorn SG, Proia AD, Yanovitch TL. et al. Ocular and histologic findings in a series of children with infantile pompe disease treated with enzyme replacement therapy. J Pediatr Ophthalmol Strabismus 2014; 51: 355-362
- 223 Slingerland NW, Polling JR, van Gelder CM. et al. Ptosis, extraocular motility disorder, and myopia as features of pompe disease. Orbit 2011; 30: 111-113
- 224 Winter AW, Salimi A, Ospina LH. et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol 2019; 103: 315-326
- 225 Yanovitch TL, Banugaria SG, Proia AD. et al. Clinical and histologic ocular findings in pompe disease. J Pediatr Ophthalmol Strabismus 2010; 47: 34-40
- 226 Wabbels B, Ali N, Kunz WS. et al. [Chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome : interdisciplinary diagnosis and therapy]. Ophthalmologe 2008; 105: 550-556
- 227 Pfeiffer MJ. [Chronic Progressive External Ophthalmoplegia Ptosis: Problems with Diagnostics and Treatment]. Klin Monbl Augenheilkd 2018; 235: 31-33
- 228 Pitceathly RD, Morrow JM, Sinclair CD. et al. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol 2016; 26: 130-137
- 229 Rahman S, Blok RB, Dahl HH. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996; 39: 343-351
- 230 McKelvie P, Infeld B, Marotta R. et al. Late-adult onset Leigh syndrome. J Clin Neurosci 2012; 19: 195-202
- 231 Lake NJ, Compton AG, Rahman S. et al. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann Neurol 2016; 79: 190-203
- 232 Han J, Lee YM, Kim SM. et al. Ophthalmological manifestations in patients with Leigh syndrome. Br J Ophthalmol 2015; 99: 528-535
- 233 Fichter N, Schittkowski M, Guthoff R. Erkrankungen der Tränendrüse. Ophthalmologe 2005; 102: 399-425
- 234 Moreiras J. Tumors of the lacrimal gland - Orbit – examination, diagnosis, mircrosurgery, pathology. 2004
- 235 Font R, Gamel J. Epithelial tumours of the lacrimal gland: an analysis of 265 cases.In Jakobiec F (ed) Ocular and adnexal tumours. Aesculapius, Birmingham. 1978
- 236 Riedel K, Markl A, Hasenfratz G. et al. Epithelial tumors of the lacrimal gland: Clinico-pathologic correlation and management. Neurosurg Rev 1990; 13: 289-298
- 237 Andreasen S, Esmaeli B, Holstein L. et al. An Update on Tumors of the Lacrimal Gland. Asia-Pac. J Ophthalmol 2017; 6: 159-172
- 238 Cates C, Manners R, Rose G. Pleomorphic adenoma of the lacrimal gland in a 10 year old girl. Br J Ophthalmol 2002; 86: 249-250
- 239 Faktorovich E, Crawford J, Char D. Benign mixed tumor (pleomorphic adenoma) of the lacrimal gland in a 6-year-old boy. Am J Ophthalmol 1996; 122: 446-447
- 240 Mercado G, Gunduz K, Shields C. et al. Pleomorphic adenoma of the lacrimal gland in a teenager. Arch Ophthalmol 1998; 116: 962-963
- 241 Ni C, Kuo P, Dryja T. Histopathological classification of 272 primary epithelial tumors of the lacrimal gland. Chin Med J 1992; 105: 481-485
- 242 Rose G, Wright J. Pleomorphic adenoma of the lacrimal gland. Br J Ophthalmol 1992; 76: 395-400
- 243 Stefko S, DiBernardo C, Green W. et al. Pleomorphic adenoma of the lacrimal gland with extensive calcification. Arch Ophthalmol 2004; 122: 778-780
- 244 Stupp T, Pavlidis M, Buchner T. Pleomorphic Adenoma of the Lacrimal Gland in a Child After Treatmentof Acute Lymphoblastic Leukemia. Arch Ophthalmol 2004; 122: 1538-1540
- 245 Bonavolontà G, Tranfa F, Staibano S. et al. Warthin Tumor of the Lacrimal Gland. Am J Ophthalmol 1997; 124: 857-858
- 246 Ghosh D, Ekka J. Adenolymphoma of the lacrimal gland: A very rare case report. Int J Ocul Oncol oculoplasty 2018; 4: 144-145
- 247 Bartoletti-Stella A, Salfi N, Ceccarelli C. et al. Mitochondrial DNA Mutations in Oncocytic Adnexal Lacrimal Glands of the Conjunctiva. ARCH OPHTHALMOL 2011; 129: 664-666
- 248 Calle C, Castillo I, Eagle R. et al. Oncocytoma of the Lacrimal Gland: Case Report and Review of the Literature. Orbit 2006; 25: 243-247
- 249 Cameron J, Barras C. Oncocytoma of the eyelid. ACTA OPHTHALMOL SCAND 2004; 83: 125
- 250 George E, Swanson P, Newman B. et al. Oculocutaneous oncocytic tumors: clinicopathologic and immunohistochemical study of 2 cases with literature review. Am J Dermatopathol 2007; 29: 279-285
- 251 Morand B, Bettega G, Bland V. et al. Oncocytoma of the eyelid: an aggressive benign tumor. Ophthalmology 1998; 105: 2220-2224
- 252 Shields C, Shields J. Tumors of the conjunctiva and cornea. Survey of Ophthalmology 2004; 49: 3-24
- 253 Shields C, Shields J, Arbizo V. Oncocytoma of the caruncle. Am J Ophthalmol 1986; 102: 315-319
- 254 Surakiatchanukul T, Sioufi K, Pointdujour-Lim R. et al. Caruncular Oncocytoma Mimicking Malignant Melanoma. Ocul Oncol Pathol 3: 320-323
- 255 Bolzoni A, Pianta L, Farina D. et al. Benign myoepithelioma of the lacrimal gland: report of a case. Eur Arch Otorhinolaryngol 2005; 262: 186-188
- 256 Font R, Garner A. Myoepithelioma of the lacrimal gland: report of a case with spindle cell morphology. Brit J Ophthalmol 1992; 76: 634-636
- 257 Grossniklaus H, Wojno T, Wilson M. et al. Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1997; 115: 1588-1590
- 258 Heathcote J, Hurwitz J, Dardick I. A Spindle-Cell Myoepithelioma of the Lacrimal Gland. Arch Ophthalmol 1990; 108: 1135-1139
- 259 Nickol T, E R Kleinsasser O. Myoepitheliome der Parotis. Jahresversammlung der Deutschen Gesellschaft für HNO-Heilkunde. 1998
- 260 Bajaj M, Pushker N, Kashyap S. Cystadenoma of the lacrimal gland. Orbit 2002; 21: 301-305
- 261 Bernardini F, Devoto M. JO C. Epithelial tumors of the lacimal gland: an update. Curr Opin in Ophthalmol 2008; 19: 409-413
- 262 Silbert J, Braich P, Misra R. Basal cell cystadenoma of the lacrimal gland: diagnostic pitfalls of a basaloid pattern in lacrimal tumours. Internat Ophthalmol. 2011 DOI: 43-46
- 263 Wang L, Zhang SK, Ma Y. et al. Papillary cystadenoma of the parotid gland: A case report. World J Clin Cases 2019; 7: 366-372
- 264 Pfeiffer M, Yin V, Bell D. et al. Sclerosing Polycystic Adenosis of the Lacrimal Gland. Ophthalmology 2013; 120: 873
- 265 Speight P, Barrett A. Salivary gland tumours: diagnostic challenges and an update on the latest WHO classification. Diagn Histopathol 2020; 26: 147-158
- 266 Brunnmann R, Ro J, Ordonez N. et al. Extrapleural Solitary Fibrous Tumor: A Clinicopathologic Study of 24 Cases. Mod Pathol 1999; 12: 1034-1042
- 267 Mupas-Uy J, Kitaguchia Y, Takahashia Y. et al. Solitary Fibrous Tumor in the Lacrimal Gland Fossa: A Case Report. Case Rep Ophthalmol 2016; 7: 398-403
- 268 Okike N, Bernatz P, Woolner L. Localized mesothelioma of the pleura. Benign and malignant variants. J Thorac Cardiovasc Surg 1978; 75: 363-372
- 269 Scott I, Tanenbaum M, Rubin D. Solitary fibrous tumor of the lacrimal gland fossa. Ophthalmology 1996; 103: 1613-1618
- 270 Vallat-Decouvelaere A, Dry S, Fletcher C. Atypical and malignant solitary fibrous tumor in extrathoracic locations; evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol 1998; 22: 1501-1511
- 271 Woo K, Suh Y, Kim Y. Solitary fibrous tumor of the lacrimal sac. Ophthalmic Plast Reconstr Surg 1999; 15: 450-453
- 272 Bielory BP, Turbin RE, Mirani NM. et al. Radiographic and histological findings in an atypical orbital myxoma. Ophthalmol Eye Dis 2011; 3: 55
- 273 Demirci H, Shields CL, Eagle RCJ. et al. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006; 124: 735-738
- 274 Kini Rao A, Nayal B. Conjunctival myxoma-a case report. Malays J Med Sci 2013; 20: 92-94
- 275 Parikh D, Mukherjee B. Lacrimal gland myxoma. Indian J Ophthalmol 2017; 65: 887-889
- 276 Rambhatla S, Subramanian N, Gangadhara Sundar JK. et al. Myxoma of the orbit. Indian J Ophthalmol 2003; 51: 85-87
- 277 Bajaj M, Pushker N, Fibrous SK. histiocytoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 145-147
- 278 Andrew N, Coupland S, Pirbhai A. et al. Lymphoid hyperplasia of the orbit and ocular adnexa: A clinical pathologic review. Survey of Ophthalmology 2016; 61: 778-790
- 279 Shields J, Shields C, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: the 2002 Montgomery lecture, part 1. Ophtalmology. 2004 DOI: 997-1008
- 280 Wittekind C. TNM-Klassifikation maligner Tumoren. Achte Auflage. Aufl. WILEY-VCH; 2017
- 281 Devoto M, Croxatto J. Primary cystadenocarcinoma of the lacrimal gland. Ophthalmology 2003; 110: 2006-2010
- 282 Takahira M, Nakamura Y, Shimizu S. et al. Carcinosarcoma of the Lacrimal Gland Arising From a Pleomorphic Adenoma. Am J Ophthalmol 2005; 140: 340-341
- 283 Daisuke Y, Toshiyuki O, Shuichi Y. Case of primary neuroendocrine carcinoma in lacrimal gland. Acta Ophthalmol 2013; 91: 252
- 284 Gess A, Silkiss R. A merkel cell carcinoma of the lacrimal gland. Ophthal Plast Reconstr Surg. 28. 11-13
- 285 El-Naggar A, Chan J, Grandis J. World Health organization classification of head and neck tumours. 2017 DOI:
- 286 Weis E, Rootman J, Joly T. et al. Epithelial lacrimal gland tumors: pathologic classification and current understanding. Arch Ophthalmol 2009; 127: 1016-1028
- 287 Gamel J, Font R. Adenoidcystic carcinoma of the lacrimal gland: the clinical significance of a basaoid histologic pattern. Human Pathol 1982; 13: 219-225
- 288 Friedrich R, Bleckmann V. Adenoid cystic carcinoma of the salivary and lacrimal glands origin: localization, classification, clinical pathological correlation, treatment results and long-term follow-up control in 84 patients. Anticancer Res 2003; 23: 937-940
- 289 Goldberg R. Intraarterial chemotherapy. A welcome new idea for the management of adenoidcystic carcinoma of the lacrimal gland. Arch Ophthalmol 1998; 116: 372-373
- 290 Hamper K, Lazar F, Dietel M. Prognostic factors for adenoid cystic carcinoma of the head and neck: a retrospectiv evaluation of 96 cases. J Oral Pathol Med 1990; 19: 101-107
- 291 Henderson J. In Orbital Tumors New York: Raven Press;. 1994: 323-342
- 292 Lee D, Campbell R, Waller R. A clinicopathologic study of primary adenoid cystic carcinoma of the lacrimal gland. Ophthalmology 1985; 92: 128-134
- 293 Strianese D, Baldi G, Staibano S. Expression of apoptosis-related markers in malignant epithelial tumors of the lacrimal gland and their relation to clinical outcome. Br J Ophthalmol 2007; 91: 1239-1243
- 294 Tse D, Benedetto P, Dubovy S. Clinical analysis oft he effect on intraarterial cytoreductive chemotherapy in the treatment of lacrimal gland andoid cystic carcinoma. Am J Ophthalmol 2006; 141: 44-53
- 295 Wright J, Rose G, Garner A. Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 1992; 76: 401-407
- 296 Karcioglu Z. Orbital Tumors. Diagnosis and Management. New York: Springer; 2005
- 297 Ahmad S, Esmaeli B, Williams M. American Joint Committee on Cancer classification predicts outcome of patients with lacrimal gland adenoid cystic carcinoma. Ophthalmology 2009; 116: 1210-1215
- 298 GROUP IHANS. Cervical lymph node metastasis in adenoid cystic carcinoma of the sinonasal tract, nasopharynx, lacrimal glands and external auditory canal: a collective international review. J Laryngol Otol 2016; 130: 1093-1097
- 299 Xiao R, Sethi R, Feng A. et al. The role of elective neck dissection in patients with adenoid cystic carcinoma of the head and neck. Laryngoscope 2019; 129: 2094-2104
- 300 Bördlein I. Mit schweren Ionen gegen hartnäckige Tumoren. Deutsches Ärzteblatt 2004; 101: 1718-1720
- 301 Hayashi K, Koto M, Ikawa H. et al. Efficacy and safety of carbon-ion radiotherapy for lacrimal gland carcinomas with extraorbital extension: a retrospective cohort study. Oncotarget 2018; 9: 12932-12940
- 302 Lesueur P, Rapeaud E, De Marzi L. et al. Adenoid Cystic Carcinoma of the Lacrimal Gland: High Dose Adjuvant Proton Therapy to Improve Patients Outcomes. Front Oncol 2020; 10: 135
- 303 Wolkow N, Jakobiec F, Lee H. et al. Long-term Outcomes of Globe-Preserving Surgery With Proton Beam Radiation for Adenoid Cystic Carcinoma of the Lacrimal Gland. Am J Ophthalmol. 2018 195.
- 304 Shields J, Shields C, Freire J. Plaque radiotherapy for selected orbital malignancies: preliminary observations: hte 2002 Montgomery Lecture, part 2. Ophthal Plast Reconstr Surg 2003; 19: 91-95
- 305 Tyl J, Blank L, Koornneef L. Brachytherapy in orbital tumors. Ophthalmology 1997; 104: 1475-1479
- 306 Gündüz A, Yesiltas Y, Shileds C. Overview of benign and malignant lacrimal gland tumors. Curr Opin Ophthalmol 2018; 29: 458-468
- 307 Ashton N. Epithelial tumors of the lacrimal gland. Mod Probl Ophthalmol 1975; 14: 306-323
- 308 Perzin K, Jakobiec F, Lovolsi V. Lacrimal gland malignant mixed tumors (carcinomas arising in benign mixed tumors): a clinicopathologic study. Cancer 1980; 45: 2593-2606
- 309 Shields C, Shields J, Eagle R. Clinicopathologic review of 142 cases of lacrimal gland lesions. Ophthalmology 1989; 96: 431-435
- 310 Henderson J, Farrow G. Primary malignant mixed tumors of the lacrimal gland. Report of 10 cases. Ophthalmology 1980; 87: 466-475
- 311 Jakobiec F, Bilyk J, Font R. Ophthalmic Pathology. An Atlas and Textbook. 4th ed. Aufl. Philadelphia. WB Saunders; 1996
- 312 Chen A, Garcia J, Bucci M. et al. The role of postoperative radiation therapy in carcinoma ex pleomorphic adenoma of the parotid gland. Int J Radiat Oncol Biol Phys 2007; 67: 138-143
- 313 Takahira M, Minato H, Takahashi M. Cystic carcinoma ex pleomorphic adenoma of the lacrimal gland. Ophthal Plast Reconstr Surg 2007; 23: 407-409
- 314 Hwang S, Kim K. High-grade Mucoepidermoid Carci- noma of the Lacrimal Gland. Korean J Ophthalmol 2018; 32: 426-427
- 315 Heaps R, Miller N, Albert D. Primary adenocarcinoma of the lacrimal gland. Ophtalmology 1993; 100: 1856-1860
- 316 von Holstein S, Coupland S. , D B. Epithelial Tumors of the Lacrimal Gland: a clinical, histopathological, surgical, and oncological survey. Acta Ophthalmol 2013; 91: 195-206
- 317 Dennie T. Metastastic her-2 amplified lacrimal gland carcinoma with response to lapatinib treatment. Case Rep Oncol Med 2015; 2015: 1-3
- 318 Totuk O, Demir M, Yapicier O. et al. Low-Grade Mucoepidermoid Carcinoma of the Lacrimal Gland in a Teenaged Patient. Case Rep Ophthalmol Med. 2017
- 319 Eviatar J, Hornblass A. Mucoepidermoid carcinoma of the lacrimal gland: 25 cases and a review and update of the literature. Ophthal Plast Reconstr Surg 1993; 9: 170-181
- 320 Sofinski S, Brown B, Rao N. Mucoepidermoid carcinoma of the lacrimal gland. Case report and review of the literature. Ophthalmic Plast Reconstr Surg 1986; 2: 147-151
- 321 Thorvaldsson S, Beahrs O, Woolner L. et al. Mucoepidermoid tumors of the major salivary glands. Am J Surg 1970; 120: 432-438
- 322 Katz E, Rootman J, Dolman P. et al. Primary Ductal Adenocarcinoma of the Lacrimal Gland. Ophthalmology 1996; 103: 157-162
- 323 Yang H, Wu C, Tsai C. et al. Primary ductal adenocarcinoma of lacrimal gland: Two case reports and review of the literature. Taiwan J Ophthalmol 2018; 8: 42-48
- 324 Ishida M, Hotta M, Kushima R. et al. Case of ductal adenocarcinoma ex pleomorphic adenoma of the lacrimal gland. Rinsho Byori 2009; 57: 746-751
- 325 Andreasen S, Grauslund M, Heegaard S. Lacrimal gland ductal carcinomas: clinical, morphological and genetic characterization and implications for targeted treatment. Acta Ophthalmol 2017; 95: 299-306
- 326 Jaspers H, Verbist B, Schoffelen R. et al. Androgen Receptor–Positive Salivary Duct Carcinoma: A Disease Entity With Promising New Treatment Options. J Clin Oncol 2011; 29: 473-476
- 327 See T, Stålhammar G, Tang T. et al. Primary ductal adenocarcinoma of the lacrimal gland: A review and report of five cases. Survey of Ophthalmology 2020; 65: 371-380
- 328 Anesi A, Negrello S, Lucchetti D. et al. Clinical Management of Acinic Cell Carcinoma of the Lacrimal Gland. Case Rep Oncol 2019; 12: 777-790
- 329 De Rosa G, Zeppa P, Tranta F. Acinic Cell Carcinoma in a Lacrimal Gland: first case report. Cancer 1986; 57: 1988-1991
- 330 Jang J, Kie J, Lee S. Acinic cell carcinoma of the lacrimal gland with intracranial extension. Ophthalmic Plast Reconstr Surg 2001; 17: 454-457
- 331 Rosenbaum P, Mahadevia P, Goodman L. Acinic cell carcinoma of the lacrimal gland. Arch Ophthalmol 1995; 113: 781-785
- 332 Briscoe D, Mahmood S, Bonshek R. et al. Primary sebaceous carcinoma of the lacrimal gland. Br J Ophthalmol 2001; 85: 625-626
- 333 Harvey P, Parsons M, Rennie I. Primary sebaceous carcinoma of lacrimal gland: a previously unreported primary neoplasm. Eye 1994; 8: 592-595
- 334 Kiratli H, Tarlan B, Fırat P. Primary sebaceous carcinoma of the lacrimal gland. Orbit 2012; 31: 352-354
- 335 Park H, Choi S. Primary sebaceous carcinoma of lacrimal gland: A case report and review of literature. World J Clin Cases 2018; 6: 1194-1198
- 336 Yamamoto N, Mizoe J, Hasegawa A. et al. Primary sebaceous carcinoma of the lacrimal gland treated by carbon ion radiotherapy. Int J Clin Oncol 2003; 8: 386-390
- 337 Herrera G. Light microscopic, ultrastructural and immunocytochemical spectrum of malignant lacrimal and salsivary gland tumors, including malignant mixed tumors. Pathobiology 1990; 58: 312-322
- 338 Iida K, Shikishima K, Okido M. et al. A case of malignant myoepithelioma in the lacrimal gland. Nippon Ganka Gakkai Zasshi 2001; 105: 42-46
- 339 Larbcharoensub N, Pangpunyakulchai D, Aroonroch R. et al. Lacrimal myoepithelial carcinoma ex recurrent pleomorphic adenoma: A clinicopathological report and review of the literature. Mol Clin Oncol 2018; 8: 209-213
- 340 Nagao T, Sugano I, Ishida Y. Salivary gland mmalignant myoepithelioma: a clinicopathologic and immunohistochemical study of ten cases. Cancer 1998; 83: 1292-1299
- 341 Xu T, Liao Z, Tang J. et al. Myoepithelial carcinoma of the headand neck: A report of 23 casesand literature review. Cancer Treatment. Communications 2014; 2: 24-29
- 342 Mahdi Y, Azami M, Daoudi R. et al. Diagnostic pitfall: primary myoepithelial carcinoma of the lacrimal gland, case report and literature review. BMC Clinical Pathology 2018; 18: 1-7
- 343 Yu G, Ma D, Sun K. Myoepithelial carcinoma of the salivary glands: behavior and management. Chin Med J 2003; 116: 163-165
- 344 Soumarová R, Lovasová Z, Vaňková M. Radiotherapy in malignant myoepithelioma of the soft palate – case report. Case Rep Oncol 2009; 2: 116-120
- 345 Nieder C, Schneller F, Grosu A. Radiotherapy and chemotherapy for myoepithelioma of the sellar region. Strahlenther Onkol 2005; 181: 260-263
- 346 Fenton S, Srinivasan S, Harnett A. et al. Primary squamous cell carcinoma of the lacrimal gland. Eye 2003; 17: 424-425
- 347 Su G, Patipa M, Font R. Primary squamous cell carcinoma arising from an epithelium-lined cyst of the lacrimal gland. Ophthal Plast Reconstr Surg 2005; 21: 383-385
- 348 Hotta K, Arisawa T, Mito H. et al. Primary squamous cell carcinoma of the lacrimal gland. Clin Experiment Ophthalmol 2005; 33: 534-536
- 349 Bernardini F, Orcioni G, Croxatto J. Oncocytic carcinoma of the lacrimal gland in a patient with neurofibromatosis. Ophthal Plast Reconstr Surg 2010; 26: 486-488
- 350 Kalantzis G, Norris J, El-Hindy N. et al. Oncocytic adenocarcinoma of the lacrimal gland: an unusual presentation. Eye 2013; 27: 104-105
- 351 Selva D, Davis G, Dodd T. Polymorphous Low-Grade Adenocarcinoma of the Lacrimal Gland. ARCH OPHTHALMOL 2004; 122: 915-917
- 352 Bortz J, Zhang P, Eagle R. et al. Secretory Carcinoma of the Lacrimal Gland: A Rare Case Report. Ophthalmic Plast Reconstr Surg 2018; 34: 154-157
- 353 Hyrczaa M, Andreasen S, Melchiorc L. et al. Primary Secretory Carcinoma of the Lacrimal Gland: Report of a New Entity. Am J Ophthalmol 2018; 193: 178-183
- 354 Kadir S. Synovial Sarcoma involving lacrimal gland: A rare clinical entity. Int J Ocu Oncol Oculoplast. 1: 14–18
- 355 Likhvantseva V, Safonova T, Kuzmin K. Primary granulocytic sarcoma of lacrimal gland. Vestn Oftalmol 2016; 132: 82-89
- 356 Yilmaz A, Saydam G, Sahin F. et al. Granulocytic sarcoma: a systematic review. Am J Blood Res 2013; 3: 265-270
- 357 Rajabi M, Riazi H, Hosseini S. et al. Malignant Peripheral Nerve Sheath Tumor of Lacrimal Nerve: A Case Report. Orbit 2015; 34: 41-44
- 358 Stark A, Hugo H, Buhl R. et al. Tumoren peripherer Nerven. Dtsch Arztebl 2002; 99: 928-933
- 359 Gündüz K, Shields J, Eagle R.. Malignant Rhabdoid Tumor of the Orbit. Arch Ophthalmol 1998; 116: 243-246
- 360 Johnson L, Sexton F, Goldberg S. Poorly differentiated primary orbital sarcoma (presumed malignant rhabdoid tumor): radiologic and histopathologic correlation. Arch Ophthalmol 1991; 109: 1275-1278
- 361 Orphanet. Rhabdoidtumor. In: Orphanet; 2020
- 362 Rootman J, Damji K, Dimmick J. Malignant rhabdoid tumor of the orbit. Ophthalmology 1989; 96: 1650-1654
- 363 Walford N, Deferrai R, Slater R. Intraorbital rhabdoid tumour following bilateral retinoblastoma. Histopathology 1992; 20: 170-173
- 364 Niffenegger J, Jakobiec F, Shore J. et al. Adult extrarenal rhabdoid tumor of the lacrimal gland. Ophthalmology 1992; 99: 567-574
- 365 Ferry J, Fung C, Zukerberg L. et al. Lymphoma of the ocular adnexa: A study of 353 cases 2007; 31: 170-184
- 366 Rasmussen P, Ralfkiaer E, Prause J. Malignant lymphoma of the lacrimal gland. A nation-based study. Arch Ophthalmol 2011; 129: 1275-1280
- 367 Johnson T, Tse D, Byrne G. Ocular adnexal lymphoid tumors: a clinicopathologic and molecular genetic study of 77 patients. Ophthal Plast Reconstr Surg 1999; 15: 171-179
- 368 Polito E, Galieni P, Leccisotti A. Clinical and radiological presentation of 95 orbital lymphoid tumors. Graefes Arch Clin Exp Ophthalmol 1996; 234: 504-509
- 369 Aranow M. Ocular adnexal lymphoma: evidence-based treatment approach. Int Ophthalmol Clin 2015; 55: 97-109
- 370 König L, Stade R, Rieber J. Radiotherapy of indolent orbital lymphomas: two radiation concepts. Strahlenther Onkol 2016; 192: 414-421
- 371 de Leval L, Jaffe E. Lymphoma Classification. Cancer J 2020; 26: 176-185
- 372 Shields J, Shields C, Eagle R. Metastatic renal cell carcinoma to the palpebral lobe of the lacrimal gland. Ophthal Plast Reconstr Surg 2001; 17: 191-194
- 373 Fowler J, Terplan K. Fibroma of the Orbit. Arch Ophthalmol 1942; 28: 263-271
- 374 Mortada A. Fibroma of the orbit. Brit J Ophthal 1971; 55: 350
- 375 Rong A, Ulloa-Padilla J, Blessing N. et al. Subperiosteal fibroma of the orbit. Orbit 2018; 37: 378-380
- 376 Rabelo N, da Silva V, do Espírito Santo M. et al. Orbit ossifying fibroma – Case report and literature review. Surg Neurol Int 2020; 11: 35
- 377 Sheikh BY. Cranial aneurysmal bone cyst “with special emphasis on endovascular management”. Acta Neurochir (Wien) 1999; 141: 601-610 discussion 610-601
- 378 Glien A, Meyer B, Wernert N. et al. [Orbital aneurysmal bone cyst in a 2-year-old child]. HNO 2007; 55: 281-286
- 379 Wang JC, Zhang M, Zhao XX. Aneurysmal bone cyst of the orbit. Chin Med J (Engl) 2015; 128: 562-563
- 380 Yu JW, Kim KU, Kim SJ. et al. Aneurysmal bone cyst of the orbit : a case report with literature review. J Korean Neurosurg Soc 2012; 51: 113-116
- 381 Ruiter DJ, van Rijssel TG, van der Velde EA. Aneurysmal bone cysts: a clinicopathological study of 105 cases. Cancer 1977; 39: 2231-2239
- 382 Biesecker JL, Marcove RC, Huvos AG. et al. Aneurysmal bone cysts. A clinicopathologic study of 66 cases. Cancer 1970; 26: 615-625
- 383 Schajowicz F. Tumors and tumorlike lesions of bone 2. Aufl. Berlin-Heidelberg-New York: Springer-Verlag; 1994
- 384 Oliveira AM, Chou MM. USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum Pathol 2014; 45: 1-11
- 385 Fay A, Dolman PJ. Laboratory Serologic Investigations.In Fay A, Dolman PJ , Hrsg. Diseases and Disorders of the Orbit and Ocular Adnexa E-book. Elsevier; 2017: 314
- 386 Mallen JR, Caranfa JT, Zimmerman D. et al. Atypical presentation of a solid-variant orbital aneurysmal bone cyst with a literature review. Neurochirurgie 2018; 64: 431-433
- 387 Chawla B, Khurana S, Kashyap S. Giant cell reparative granuloma of the orbit. Ophthalmic Plast Reconstr Surg 2013; 29: e94-95
- 388 Kamoshima Y, Sawamura Y, Imai T. et al. Giant cell tumor of the frontal bone in a girl: case report. Neurol Med Chir (Tokyo) 2011; 51: 798-800
- 389 Smolarz-Wojnowska A, Essig H, Gellrich NC. et al. [Orbital tumours in children and adolescents. Differential diagnostics and clinical symptoms]. Ophthalmologe 2010; 107: 543-548
- 390 Vernet O, Ducrey N, Deruaz JP. et al. Giant cell tumor of the orbit. Neurosurgery 1993; 32: 848-851
- 391 Yip CM, Lee HP, Hsu SS. et al. Left orbital roof giant cell tumor of bone: A case report. Surg Neurol Int 2018; 9: 127
- 392 Yamaguchi T, Dorfman HD. Giant cell reparative granuloma: a comparative clinicopathologic study of lesions in gnathic and extragnathic sites. Int J Surg Pathol 2001; 9: 189-200
- 393 de Lange J, van den Akker HP, van den Berg H. Central giant cell granuloma of the jaw: a review of the literature with emphasis on therapy options. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2007; 104: 603-615
- 394 Auclair PL, Cuenin P, Kratochvil FJ. et al. A clinical and histomorphologic comparison of the central giant cell granuloma and the giant cell tumor. Oral Surg Oral Med Oral Pathol 1988; 66: 197-208
- 395 de Lange J, van Rijn RR, van den Berg H. et al. Regression of central giant cell granuloma by a combination of imatinib and interferon: a case report. Br J Oral Maxillofac Surg 2009; 47: 59-61
- 396 Roman-Romero L, Gonzalez-Garcia R. Cholesterol granuloma of the orbit. Report of cases and analysis of controversial treatment. J Maxillofac Oral Surg 2011; 10: 166-169
- 397 Loeffler KU, Kommerell G. Cholesterol granuloma of the orbit--pathogenesis and surgical management. Int Ophthalmol 1997; 21: 93-98
- 398 Selva D, Phipps SE, O'Connell JX. et al. Pathogenesis of orbital cholesterol granuloma. Clin Exp Ophthalmol 2003; 31: 78-82
- 399 Daus W, Voges J, Schwechheimer K. et al. [Cholesterol granuloma of the orbits--clinicopathologic study]. Klin Monbl Augenheilkd 1988; 193: 195-199
- 400 Hsu HT, Liao WC, Wu CC. et al. Cholesterol granuloma of the orbit. Kaohsiung J Med Sci 2017; 33: 422-423
- 401 Rong AJ, Erickson BP, Blessing NW. et al. Orbital cholesterol granuloma: A report and discussion of orbital findings. Am J Ophthalmol Case Rep 2019; 15: 100468
- 402 Arat YO, Chaudhry IA, Boniuk M. Orbitofrontal cholesterol granuloma: distinct diagnostic features and management. Ophthalmic Plast Reconstr Surg 2003; 19: 382-387
- 403 Aferzon M, Millman B, O'Donnell TR. et al. Cholesterol granuloma of the frontal bone. Otolaryngol Head Neck Surg 2002; 127: 578-581
- 404 Kabra R, Patel S, Shanbhag S. Orbital Chondroma: A rare mesenchymal tumor of orbit. Indian J Ophthalmol 2015; 63: 551-554
- 405 Garrity J, Henderson J. Henderson’s Orbital Tumors.In 4th ed. Aufl. USA: Lippincott Williams and Wilkis; 2007: 103–104
- 406 Rootman J, Connell J. Primary bone tumors of orbit. Surv Ophthalmol 2004; 49: 328-342
- 407 Shields J, Bakewell B, Augsburger J. et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984; 102: 1606-1611
- 408 Pushker N, Meel R, Sharma S. et al. Giant orbital Schwannoma with fluid-fluid-levels. Br J Ophthalmol 2010; 95: 1168
- 409 Misra S, Gogri P, Misra N. et al. Recurrent neurofibroma of the orbit. Australas Med J 2013; 30: 189-191
- 410 Lee L, Gigantelli J, Kincaid M. Localized neurofibroma of the orbit: a radiographic and histopathologic study. Ophthal Plast Reconstr Surg 2000; 16: 241-246
- 411 Turbin R, Thompson C, Kennerdell J. et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophtalmology 2002; 109: 890-899
- 412 Schittkowski M, Fichter N., Orbitale Neoplasien Teil I. Einführung, vaskuläre und neurogene Neoplasien. Ophthalmologe 2012; 109: 1033-1046
- 413 Jain D, Ebrahimi K, Miller N. et al. Intraorbital meningiomas: a pathologic review using current World Health Organization criteria. Arch Pathol Lab Med 2010; 134: 766-770
- 414 Eddleman C, Liu J. Optic nerve sheath meningioma: current diagnosis and treatment. Neurosurg Focus 2007; 23: E4
- 415 Saeed P, Rootman J, Nugent R. et al. Optic nerve sheath meningiomas. Ophthalmology 2003; 110: 2019-2030
- 416 Ramey W, Arnold S, Chiu A. et al. A Rare Case of Optic Nerve Schwannoma: Case Report and Review of the Literature. Cureus 2015; 7: e265
- 417 Gnekow A. Low-grade chiasmatic-hypothalamic glioma-carboplatin and vincristine chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy - report from the multicenter treatment study for children and adolescents with a low grade glimoa - HIT-LGG 1996 - of the Society of Paediatric Oncology and Hematology (GPOH). Klin Padiatr 2004; 216: 331-342
- 418 Sharif S. Second primary tumors in neurofibromatosis 1 patients treated for optic nerve glioma: substantial risks after radiotherapy. J Clin Oncol 2006; 24: 2570-2575
- 419 Wilhelm H. Primary optic nerve tumors. Curr Opin Neurol 2009; 22: 11-18
- 420 Emesz M, Arlt E, Krall E. et al. Granularzelltumor der Orbita. Der. Ophthalmologe 2014; 111: 866-870
- 421 Huang N, Rayess H, Svider P. et al. Orbital Paraganglioma: A Systematic Review. J Neurol Surg B Skull Base 2017; 79: 407-412
- 422 Makhdoomi R, Nayil K, Santosh V. et al. Orbital paraganglioma--a case report and review of the literature. Clin Neuropathol 2010; 29: 100-104
- 423 Al-Khiary H, Ayoubi A, Elkhamary S. Primary orbital ganglioneuroma in a 2-year-old healthy boy. Saudi J Ophthalmol 2010; 24: 101-104
- 424 Cannon T, Brown H, Hughes B. Orbital Ganglioneuroma in a Patient With Chronic Progressive Proptosis. Arch Ophthalmol 2004; 122: 1712-1714
- 425 Hatsukawa Y, Furukawa A, Kawamura H. et al. Rhabdomyoma of the orbit in a child. Am J Ophthalmol 1997; 123: 142-144
- 426 Li Y, Li N, Zang W. et al. Rhabdomyoma of the orbit. J Pediatr Ophthalmol Strabismus 2008; 45: 113-115
- 427 Myung J, Kim I, Chun J et al. Rhabdomyoma of the orbit: a case report. Pediatric Radiolog. y. 32: 589–592
- 428 Gündüz K, Günalp I, Erden E. et al. Orbital leiomyoma: report of a case and review of the literature. Surv Ophthalmol 2004; 49: 237-242
- 429 Merani R, Khannah G, Mann S. et al. Orbital leiomyoma: a case report with clinical, radiological and pathological correlation. Clin Exp Ophthalmol 2005; 33: 408-411
- 430 Shah N, Chang W, White V. et al. Orbital lipoma: 2 cases and review of literature. Ophthalmic Plast Reconstr Surg 2007; 23: 202-205
- 431 Franchi G, Sleilati E, Soupre V. et al. Melanotic neuroectodermal tumour of infancy involving the orbit and maxilla: surgical management and follow-up strategy. Br J Plast Surg 2002; 55: 526-529
- 432 Koob M, Fayard C, Pariente D. et al. Prenatal diagnosis of orbital melanotic neuroectodermal tumor in infancy. Ultrasound Obstet Gynecol 2015; 46: 247-252
- 433 Soles B, Wilson A, Lucas D. et al. Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 2018; 142: 1358-1363
- 434 NIH. Childhood Rhabdomyosarcoma Treatment (PDQ®)–Health Professional Version. In: National Cancer Institute at the National Institutes of Health; 2020
- 435 Al-Bargi H, Al-Maiman M. Orbital fibrosarcoma: a case report and literature review. Egypt J Oral Maxillofac Surg 2012; 3: 45-48
- 436 Bahrami A, Folpe A. Adult-type fibrosarcoma: A reevaluation of 163 putative cases diagnosed at a single institution over a 48-year period. Am J Surg Pathol 2010; 34: 1504-1513
- 437 Scruggs B, Ho S, Valenzuela A. Diagnostic challenges in primary orbital fibrosarcoma: a case report. Clin Ophthalmol 2014; 8: 2319-2323
- 438 Almater A, Alfaleh A, Alshomar K et al. Retinoblastoma: Update on Current Management, Retinoblastoma - Past, Present and Future. In Alkatan H, Hrsg.Retinoblastoma: IntechOpen; 2019
- 439 Cassoux N, Lumbroso L, Levy-Gabriel C. et al. Retinoblastoma: Update on Current Management. Asia-Pac. J Ophthalmol 2017; 6: 290-295
- 440 WHO. RETINOBLASTOMA - 2014 Review of Cancer Medicines on the WHO List of Essential Medicines. In: Union for International Cancer Control; 2014
- 441 Caignard A, Faguer R, Mercier P. et al. Optic nerve and visual pathways primary glioblastoma treated with radiotherapy and temozolomide chemotherapy. Eur J Ophthalmol 2014; 24: 637-640
- 442 Waheed S, Huang B, Zamora C. Glioblastoma involving the optic nerve. Am J Neuroradiol 2018; 41: e
- 443 Alireza M, Amelot A, Chauvet D. Poor prognosis and challenging treatment of optic nerve malignant gliomas: Literature review and case report series. World Neurosurg 2017; 97: e1-e6
- 444 Ashour O, Stalling M, Ramsey J. et al. Intraocular Medulloepithelioma. RadioGraphics 2018; 38: 194-199
- 445 Verdijk R. On the Classification and Grading of Medulloepithelioma of the Eye. Ocul Oncol Pathol 2016; 2: 190-193
- 446 Hei Y, Kang L, Yang X. Orbital alveolar soft part sarcoma: A report of 8 cases and review of the literature. Oncol Lett 2018; 15: 304-314
- 447 Inci E, Korkut N, Erem M. et al. [Alveolar soft tissue sarcoma]. HNO 2004; 52: 145-149
- 448 Rose A, Kabiru J, Rose G. Alveolar soft-part sarcoma of the orbit. Afr J Paediatr Surg 2011; 8: 82-84
- 449 Dirksen U, Jürgens H. S1-Leitlinie: Ewing-Sarkome des Kindes- und Jugendalters. Stand 06/2014, DOI: AWMF-Register Nr. 025/006
- 450 Kaliki S, Rathi S, Palkonda V. Primary orbital Ewing sarcoma family of tumors: a study of 12 cases. Eye 2018; 32: 615-621
- 451 Chaugule S, Putambekar A, Gavade S. et al. Primary Orbital Leiomyosarcoma in an Adult Male. Ophthal Plast Reconstr Surg 2019; 35: e27-e29
- 452 Klippenstein K, Wesley R, Glick A. Orbital Leiomyosarcoma After Retinoblastoma. Ophthalmic Surg Lasers Imaging. Retina 1999; 30: 579-583
- 453 Yeniad B, Tuncer S, Peksayar G. et al. Primary Orbital Leiomyosarcoma. Ophth. Plast Reconstr Surg 2009; 25: 154-155
- 454 Cai Y, McMenamin E, Geoff R. et al. Primary liposarcoma of the orbit: A clinicopathologic study of seven cases. Ann Diagn Pathol 2001; 5: 255-266
- 455 Garrity J, Henderson J, Cameron J. Henderson's Tumors of Primitive Mesoderm, Striated muscle, Adipose Tissue and Smooth muscle.In Garrity JA HJ, Cameron JD , Hrsg. Henderson's orbital tumors. 4th edition. AuflPhiladelphia: Lippincott Williams and Wilkins; 2007: 123–141
- 456 Alkatan H, Chaudhry I, Al-Qahtani A. Epithelioid sarcoma of the orbit. Ann Saudi Med 2011; 31: 187-189
- 457 Inazawa N, Hatakeyama N, Hori T. et al. Primary orbital neuroblastoma in a 1-month-old boy. Pediatr Int 2014; 56: 122-125
- 458 Vallinayagam M, Rao V, Pandian D. et al. Primary orbital neuroblastoma with intraocular extension. Indian J Ophthalmol 2015; 63: 684-686
- 459 Zhang N, Lin L. Presumed primary orbital neuroblastoma in a 20-month-old female. Ophthal Plast Reconstr Surg 2010; 26: 383-385
- 460 Rose A, Luthert P, Jayasena C. et al. Primary Orbital Melanoma: Presentation, Treatment, and Long-term Outcomes for 13 Patients. Front Oncol 2017; 7: 316
- 461 Kiratli H, Balci K, Güler G. Primary orbital endodermal sinus tumor (yolk sac tumor). J AAPOS 2008; 12: 623-625
- 462 Mogaddam A, Memar B, Aledavood A. et al. Isolated Orbital Endodermal Sinus Tumor. Ophthal Plast Reconstr Surg 2007; 23: 477-479
- 463 Eckardt A, Lemound J, Rana M. et al. Orbital lymphoma: diagnostic approach and treatment outcome. World J Surg Oncol. 2013 DOI: 73
- 464 Olsen T, Heegaard S. Orbitla Lymphoma. Surv Ophthalmol 2019; 64: 45-66
- 465 Sharma T, Kamath M. Diagnosis and Management of Orbital Lymphoma. Ophthalmic Pearls. 2015 DOI: 37-39
- 466 Bidar M, Wilson M, Laquis S. et al. Clinical and imaging characteristics of orbital leukemic tumors. Ophthalmic Plast Reconstr Surg 2007; 23: 87-93
- 467 Hatton M, Rubin P. Chronic Lymphocytic Leukemia of the Orbit. Arch Ophthalmol 2002; 120: 990-991
- 468 Maka E, Lukáts O, Tóth J. et al. Orbital tumour as initial manifestation of acute myeloid leukemia: granulocytic sarcoma: case report. Pathol Oncol Res 2008; 14: 209-211
- 469 Taylor C, Taylor R, Kinsey S. LEUKEMIC INFILTRATION OF THE ORBIT: Report of Three Cases and Literature Review. Pediatr Hematol Oncol 2005; 22: 415-422
- 470 Pausch N, Sterker I, Bauer U. Maligner intraorbitaler Tumor: Primum oder Metastase?. HNO 2016; 64: 262-264
- 471 Schenke T, Lehmann G, Kunze C. et al. Eine abszessverdächtige Raumforderung in der extraokulären Augenmuskulatur. HNO. 2020
- 472 Suarez C, Ferlito A, Lund V. Management of the orbit in malignant sinunasal tumors. Head Neck 2008; 30: 242-250
- 473 Evert K, Kühnel T, Weiß K. et al. Diagnostik und Management von Gefäßanomalien. Der. Pathologe 2019; 40: 422-430
- 474 Müller-Wille R, Wildgruber M, Sadick M. et al. Vascular Anomalies (Part II): Interventional Therapy of Peripheral Vascular Malformations. Fortschr Röntgenstr 2018; 190: 927-937
- 475 Sadick M, Müller-Wille R, Wildgruber M. et al. Vascular Anomalies (Part I): Classification and Diagnostics of Vascular Anomalies. Fortschr Röntgenstr 2018; 190: 825-835
- 476 Wassef M, Blei F, Adams D. et al. Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 2015; 136: e203-e214
- 477 Fay A, Nguyen J, Jakobiec F. et al. Propranolol for Isolated Orbital Infantile Hemangioma. ARCH OPHTHALMOL 2010; 128: 256-258
- 478 Grantzow R, Schmittenbecher P, Höger P. et al. S2k-Leitlinie 006/100: Infantile Hämangiome im Säuglings- und Kleinkindesalter. AWMF online. 2015 DOI: 1-13
- 479 Goldsmith J, van de Rijn M, Syed N. Orbital Hemangiopericytoma and Solitary Fibrous Tumor: A Morphologic Continuum. Internat J Surg Pathol 2001; 9: 295-302
- 480 Pacheco L, Fernandes B, Miyamoto C. et al. Rapid growth of an orbital hemangiopericytoma with atypical histopathological findings. Clin Ophthalmol 2014; 8: 31-33
- 481 Westerfeld C, Allen C, Mihm M. et al. Non-Involuting Congenital Hemangioma Presenting as an Orbital Mass in a Newborn. Investigative Ophthalmology & Visual Science 48: 3582
- 482 Gbolahan O, Fasina O, Adisa A. et al. Spindle cell hemangioma: Unusual presentation of an uncommon tumor. J Oral Maxillofac Pathol 2015; 19: 406
- 483 Budimir I, Demirović A, Iveković R. et al. Epithelioid hemangioma of the orbit: case report. Acta Clin Croat 2015; 54: 92-95
- 484 McEachren T, Brownstein S, Jordan D. et al. Epithelioid hemangioma of the orbit. Ophtalmology 2000; 107: 806-810
- 485 Stiefel H, Ng J, Wilson D. et al. Orbital Cellular Epithelioid Hemangioma. Ocul Oncol Pathol 2019; 5: 424-431
- 486 Kamano H, Noguchi T, Yoshiura T. et al. Intraorbital lobular capillary hemangioma (pyogenic granuloma). Radiat Med 2008; 26: 609-612
- 487 Häußler S, Uecker F, Knopke S. et al. Tufted angiomas of the head and neck. HNO 2018; 66: S1-S6
- 488 Benesch M, Lackner H, Sorantin E. et al. Medikamentöse Therapie vaskulärer Anomalien. Paediatr Paedolog 2020; 55: 21-27
- 489 Cho W, Kim S, Park S. et al. Intracranial kaposiform hemangioendothelioma: proposal of a new malignant variant. J Pediatr Ophthalmol Strabismus 2009; 3: 147-150
- 490 Li B, Li Y, Tian X. et al. Unusual multifocal intraosseous papillary intralymphatic angioendothelioma (Dabska tumor) of facial bones: a case report and review of literature. Diagn Pathol 2013; 8: 160
- 491 Collaco L, Goncalves M, Gomes L. Orbital Kaposi’s Sarcoma in Acquired Immunodeficiency Syndrome. Eur J Ophthalmol 2000; 10: 88-90
- 492 Siddens J, Fishman J, Jackson I. et al. Primary orbital angiosarcoma: a case report. Ophthalmic Plast Reconstr Surg 1999; 15: 454-459
- 493 Lyon D, Tang T, Kidder T. Epithelioid Hemangioendothelioma of the Orbital Bones. Ophtalmology 1992; 99: 1773-1778
- 494 ISSVA. ISSVA Classification of Vascular Anomalies ©2018. In: International Society for the Study of Vascular Anomalies
- 495 Wildgruber M, Sadick M, Muller-Wille R. et al. Vascular tumors in infants and adolescents. Insights Imaging 2019; 10: 30
- 496 Gilbert P, Dubois J, Giroux F. New Treatment Approaches to Arteriovenous Malformations. Semin Intervent Radiol 2017; 34: 258-271
- 497 Schobinger RA, Fontaine R. Periphere Angiodysplasien.