
Subscribe to RSS
DOI: 10.1055/a-1331-2469
Rare Diseases of the Nose, the Paranasal Sinuses, and the Anterior Skull Base
Article in several languages: deutsch | English
- 1. Introduction
- 2 Rare Diseases of the Nose, the Paranasal Sinuses, and the Snterior Skull Base
- 2.4 Tumors
- Summary
- Literaturverzeichnis
Abstract
Due to their low incidence and thus resulting limited diagnostic criteria as well as therapeutic options, rare diseases of the nose, the paranasal sinuses, and the anterior skull base are a significant challenge. The value as of which a disease has to be considered as rare amounts to a maximum of 5 patients per 10 000 people. Within these diseases, however, there are extreme differences. Some rare or orphan diseases like for example the inverted papilloma belong to regularly diagnosed and treated diseases of larger departments of oto-rhino-laryngology whereas other rare diseases and malformations have only been described in less than 100 case reports worldwide. This fact emphasizes the necessity of bundling the available experience of diagnostics and therapy. The present article gives an overview about rare diseases of the nose, the paranasal sinuses, and the anterior skull base from the field of diseases/syndromes of the olfactory system, malformations of the nose and paranasal sinuses, ventilation and functional disorders as well as benign and malignant tumors. The classification and data on diagnostic and therapeutic options were established based on the current literature.
1. Introduction
1.1 Definition and epidemiological aspects of rare diseases
In the European Union, a disease is considered as rare when a maximum of 5 per 10 000 people are affected [1]. In many cases, orphan diseases present an important interdisciplinary challenge with regard to the correct diagnosis. Within the EU, about 6000 diseases are listed as “rare”. In Germany, about 4 million patients suffer from orphan diseases, in Europe the estimated number amounts to about 30 million. Due to the diversity of the diseases and the low number of affected people, research in diagnostics and therapy is difficult because of socio-economic reasons.
The number and definition of orphan diseases vary regionally due to epidemiological factors. Thus, for example infectious diseases in developing countries may occur frequently whereas they have a low incidence in Europe.
1.2 Definition of the terms of incidence and prevalence
The incidence describes the number of newly diagnosed cases of a certain disease in a population within a defined period of time (mostly 1 year).
The prevalence defines the total number of diseases in a population at a certain time or within a certain period of time.
Incidence=newly diagnosed cases/population
Prevalence=number of cases/population x100
For some diseases that are described in the present article, exact data on their incidence do not exist because the current literature only contains very few case reports. The website of http://www.orpha.net was founded in 1997 in France. Since 2000, it is promoted by the European Commission and provides information on many orphan diseases. The published information with regard to incidence and prevalence are based on original data collected on a worldwide or European level or extrapolated original data as long as a founder effect (deviation of an isolated population from a stem population) can be excluded for the disease [2].
1.3 Nose, paranasal sinuses, and anterior skull base
As part of the upper airways, the human nose is responsible for conditioning the inhaled air. The intact function of the mucosa consists of a mucosal film that moves continuously by means of cilia. This layer ensures the humidification of the inspired air and on the other hand it is a barrier against inhaled foreign bodies or pathogens together with the sneezing reflex [3] [4]. The regular distribution of the inhaled air over a possibly large surface of the nasal mucosa is essential for the intact function of the nose [4] [5] [6] [7]. A multitude of diseases may lead to an impairment or loss of conditioning and protection as well as of the olfactory function [6] [8].
Beside typical diseases such as chronic rhinosinusitis with nasal polyps (CRSwNP) or the viral rhinitis, also some rare diseases exist that lead to an impairment or complete loss of this function. Often they are diagnosed too late because of the similarity of the symptoms.
Due to their direct neighborhood, nose, paranasal sinuses, and anterior skull base are an anatomical unit that cannot be separately considered with regard to the diseases in this area. So there are only few diseases that affect only one of the mentioned areas. Thus, a strict separation of inflammatory and tumorous diseases as well as malformations in the areas of the nasal cavity, the paranasal sinuses, or the anterior skull base are not always possible because many pathologies concern more than one of the single sub-areas.
With an incidence of about 1–1.5:100 000, sinonasal tumors meet the criteria of an orphan disease [9] [10]. These malignant neoplasms represent only 3% of all head and neck carcinomas and less than 1% of all malignant diseases of the entire human body [11] [12] [13] [14]. Hence, all malignant entities of the nose and the paranasal sinuses as well as the anterior skull base are rare diseases according to the definition.
Tumors of the nose, the paranasal sinuses, and the anterior skull base cannot always be differentiated based on the anatomical region because of the narrow circumstances often several regions are affected even in low stages. The TNM classification of carcinomas of the nasal cavity and the paranasal sinuses is summarized in [Table 1].
|
T categories of the nasal cavity and ethmoid sinus |
|
|---|---|
|
T1 |
Tumor restricted to any 1 subsite, with or without bony invasion |
|
T2 |
Tumor invading 2 subsites in a single region or extending to involve an adjacent region within the nasoethmoidal complex, with or without bony invasion |
|
T3 |
Tumor extends to invade the medial wall or floor of the orbit, maxillary sinus, palate, or cribriform plate |
|
T4 |
Tumor invades any of the following: |
|
T4a |
Anterior orbital contents, skin of the nose or cheek, minimal extension to the anterior cranial fossa, pterygoid plates, sphenoid or frontal sinuses |
|
T4b |
Orbital apex, dura, brain, middle cranial fossa, cranial nerves other than maxillary division of trigeminal nerve (V2), nasopharynx, or clivus |
2 Rare Diseases of the Nose, the Paranasal Sinuses, and the Snterior Skull Base
Rare diseases of the nose, paranasal sinuses, and skull base may be classified into the following categories:
-
Diseases/syndromes of the olfactory system
-
Malformations
-
Ventilation and functional disorders
-
Benign and malignant tumors
-
Inflammatory/granulomatous diseases
An article from 2015 by Martin Laudien already described rare rhinological diseases focusing on a granulomatous genesis [15] so that the present contribution will concentrate on the first four aspects. Hereby, the most important rare diseases of the olfactory system, malformations, cancer diseases, and functional disorders of the nose, the paranasal sinuses, and the anterior skull base are discussed taking into account the current literature.
2.1 Diseases/syndromes of the olfactory system
The genesis of disorders of the olfactory system is manifold. Possible origins are rhinological diseases, traumas, neoplasms, and congenital disorders or they are called idiopathic [16] [17]. Furthermore, many viral diseases exist that may be accompanied by temporary or permanent hyposmia or anosmia. Due to the motto of the annual meeting, the present article does not focus on all rare diseases but only on those that concern primarily the smelling function. Those are the following syndromes and malformations:
-
Isolated congenital anosmia
-
Kallmann syndrome
-
Neuroectodermal syndrome, Johnson type
2.1.1 Isolated congenital anosmia
Isolated congenital anosmia is extremely rare. Worldwide only 15 cases of isolated congenital anosmia have been described up to now [18]. In these cases, the anosmia is already present since birth. The origin is a developmental disorder of the olfactory bulb that may occur on one or both sides [19]. Another possible genesis may be the replacement of olfactory epithelium by respiratory epithelium which lines the nasal cavity. The origin is an autosomal-dominant disorder with incomplete penetrance. There is no causal therapy for this malformation.
2.1.2 Kallmann syndrome
Kallmann syndrome describes an inherited developmental disorder, in its context, a congenital hypogonadotropic hypogonadism develops due to a gonadotropin-releasing hormone (GnRH) deficiency. In addition, affected patients suffer from hyposmia or anosmia (in cases of hypoplasia or aplasia of the olfactory bulb) that indicates an interrupted embryonic migration of the GnRH synthesizing neurons of the olfactory epithelium into the area of the hypothalamus. The inheritance is X chromosomal recessive [20]. The prevalence of Kallmann syndrome is estimated to 3.75:100,000 [2]. Therapies aim at inducing the puberty and later fertility. While in this context good therapy outcomes may be achieved, treatment of hyposmia or anosmia is not possible.
2.1.3 Johnson neuroectodermal syndrome
The neuroectodermal syndrome, Johnson type, comprises the symptoms or alopezia, anosmia or hyposmia, conductive hearing loss, malformation of the auricles, microtia and/or atresia of the external auditory canal and hypogonadotropic hypogonadism. According to the first descriptions, it is also called Johnson-McMillin syndrome [21] [22]. The inheritance occurs autosomal-dominantly, however, the exact etiology is unknown. It is assumed that an embryological defect during the differentiation of the neural crest of the head region is responsible.
The prevalence of this syndrome is estimated to clearly less than 1:1 000 000. Therapy for hyposmia or anosmia associated with this syndrome does not exist.
2.2 Malformations of the nose and paranasal sinuses
In the current literature, more than 300 syndromes involving the nose are described. In many of these syndromes, also the development of the nose may be impaired, however this pathology may be considered as subordinate in the overview of the pathologies occurring in the context of the syndrome. For reasons of clarity, the following paragraph will describe pathologies that primarily affect the nose or – in cases of severer disease – also the paranasal sinuses and the anterior skull base. The following malformations are included:
-
Arrhinia/hemirhinia
-
Bifid nose
-
Craniorhinia
-
Craniofacial clefts and paramedian cleft nose
-
Duplication anomalies of the nose
-
Lateral proboscis
-
Fistula of the nasal ridge
2.2.1 Arrhinia/hemirhinia
Congenital arrhinia is an extremely rare malformation where the nose is not or only rudimentarily developed ([Fig. 1]). Its pathogenesis is not yet completely clarified. It is assumed that a developmental disorder of the geminated nasal placodes between the 3rd and 10th week of gestation plays a role in its genesis. The early fusion of the median nasal processes, missing resorption of the nasal epithelial plug as well as an abnormal migration of cells of the neural crest are discussed as further possible mechanisms. Arrhinia may occur as singular malformation or in the context of syndromes such as the Treacher-Collins syndrome that consists of oto-mandibular dysplasia with various defects in the head and neck area and the Bosma arrhinia-microphthalmia syndrome or Bosma-Henkin-Christiansen syndrome, a combination of arrhinia, choanal atresia, and microphthalmia.

Worldwide, about 20 cases have been described [2]. Even rarer are cases of congenital unilateral arrhinia (hemirhinia, missing side of the nose [Fig. 2]). In some cases, completely missing nose with palpatorily solid base is described ([Fig. 1]), other cases reveal a rudimentary nose with blind ending hump or dimple [23].

Most cases of arrhinia that have been reported, have occurred sporadically and show an inconspicuous karyotype [24] [25]. The survival rate of patients with this malformation is rather low because the findings may lead to severe upper airway obstruction, stridor, and infections of the airways as well as malnutrition [26]. Surgery is the therapy of choice, however, only few data are available with regard to the technique and the optimal time for treatment. Tracheostomy should be performed initially in order to secure breathing. It is generally recommended to plan reconstructive measure up to the preschool age at the latest.
2.2.2 Bifid nose
The bifid nose is a rare congenital malformation that is probably autosomal-dominantly or recessively inherited. Characteristic is a nasal cleft, which presents in various manifestations. It may be a nearly unremarkable fold at the columella ([Fig. 3]) up to complete split of the underlying bone and cartilage which may lead to the development of two half noses. In the context of this malformation, the airway is usually adequately developed. Bifid nose may be considered as mild type of frontonasal dysplasia [27], but also other malformations like hypertelorism and midline cleft of the lip are observed in relation with the nasal malformation [28].

The origin of frontonasal dysplasia is unknown. Regarding the etiology, a deficient development of the nasal capsula is assumed, probably the migration of the olfactory epithelium into the nasal capsula is interrupted in the course of the 4th and 6th week of embryogenesis. The capsule cannot be fully developed and the primitive brain tissue fills the space between the dehiscent nasal ridge [29].
CT scan or MRI is urgently required prior to surgery because also mild forms of frontonasal dysplasia may be associated with intracranial anomalies.
In cases of mild types of bifid nose, surgery typically consists of open rhinoplasty [27] which allows a good overview of the site and preserves the vascular supply of the skin and the soft tissue of the nose. It is assumed that the columella incision as well as the lifting of the nasal skin applied in open technique do not influence the growth of the child’s nose [30].
2.2.3 Craniorhinia
The characteristics of craniorhinia are brachycephaly, receding forehead, and sclerotic skull base, however without craniosynostoses. The nasolacrimal duct is not developed. The configuration of the nose is broad, the ala of the nose appear distended and anteverted. A nasal hirsutism and bilaterally symmetric, round, cystic structures with small fistulas are observed directly below the nose. Hypertelorism may be found in addition. Probably, the inheritance is autosomal-dominant, however, cases with consanguine parents allow the assumption that pseudo-dominant autosomal-recessive inheritance cannot be fully excluded [31] [32]. Worldwide, four affected families have been described [2].
2.2.4 Craniofacial clefts and paramedian cleft nose
Craniofacial clefts and are extremely rare malformations of embryogenesis. Primary or true clefts occur between the 4th and 8th week of gestation because the fusion between the different facial processes could not be fully completed. Secondary or pseudo-clefts appear later. They concern the mesenchymal differentiation and may be considered as dysplasia. In both situations, the future growth potential is reduced compared to the remaining parts of the face. The incidence of craniofacial clefts amounts to 1.4–4.9:100 000 [33].
The cleft formation may concern brain tissue, soft tissue, and bones. Bony malformations occur at the front, orbita, ethmoid sinus, maxilla, and palate. Meningoceles and meningoencephaloceles may develop in cases of intracranial involvement.
Median and paramedian facial clefts are often associated with hypertelorism, anterior or basal encephalocele, positional anomaly of the maxilla and nasal deformities ([Fig. 4]). Also malformations of the soft parts such as cleft lip and palate and eyelid coloboma can present [33].

The paramedian nasal cleft is a rare developmental defect during embryogenesis that is characterized by unilateral or bilateral coloboma of the nose ([Fig. 5]). It is a mild form of craniofacial cleft. The malformation can appear as small notch leading to an unimportant deviation of the nasal septum up to nasal clefts of different size that can be associated with small cysts of the paranasal sinuses in the nasal midline. Paramedian nasal clefts occur in an isolated way or in combination with cleft lip and/or other craniofacial anomalies (e. g. hypertelorism, broad nasal root, midline cleft). Nasal ridge and tip are usually well developed [34].

2.2.5 Duplication anomalies of the nose: polyrhinia and accessory nostril
Duplication anomalies of the nose comprise polyrhinia (double nose) and the accessory nostril. Both nasal deformities occur extremely rarely (according to Orphanet, their incidence is estimated to less than 1:1 000 000). In the literature, a total of 8 cases have been described, 4 of them had polyrhinia and 3 an isolated accessory nostril as well as one patient with accessory nostril in combination with cleft lip [35].
Polyrhinia is a congenital malformation appearing as a complete duplication of the nose. All published cases were sporadic. It is assumed that the malformation is based on an embryonic developmental defect with duplication of the medial nasal process [36].
Also the accessory nostril is a very rare congenital malformation that is characterized by the presence of one or several accessory nostrils with or without accessory cartilage. The accessory nostrils are located medial, above, below, or lateral to the other nostrils. In contrast to polyrhinia, no duplication of the nasal septum is found. The accessory nostril is frequently associated with other malformations of the head and neck [37] [38].
2.2.6 Lateral proboscis
Lateral proboscis is an extremely rare malformation that was first described in 1861. The deficient side of the nose shows a trunk-like rudimentary nose that may start at every point along the embryonic fusion line between the anterior maxilla and the frontonasal process. In most cases, the location of this rudiment is at the medial part of the orbital roof [39].
The exact embryonic mechanism that is responsible for the development of lateral proboscis could not yet be clarified. The more popular theories suppose the incomplete fusion of the lateral nasal and maxillary processes and the irregular fusion of the maxillary process of the affected side with the medial nasal process [40] [41] [42].
According to existing data, it is recommended to start surgical therapy early in childhood in order to reduce or avoid psychosocial consequences. The final esthetic reconstruction of the nose, however, should be performed in later adolescence when the growth of the nasal skeleton is completed.
There are different surgical techniques for initial correction of lateral proboscis. The technique that is frequently described consists of deepithelialization of the middle and distal part of the rudimentary trunk that is then inserted into the opened and malformed ipsilateral nasal wall [39].
2.2.7 Fistula of the nasal ridge
Fistula of the nasal ridge is a rare malformation that is defined by the presence of a dermoid cyst on the nasal ridge. The incidence amounts to 1:20 000–1:40 000 [43]. Clinically, it presents as a solid palpable, slowly growing mass that contains skin and dermal elements such as hair follicles and sebaceous glands. Intermitting or chronic secretion of sebum and serous liquid as well as local infections may occur. In single cases, an intracranial connection is found so that history taking should focus on complaints of meningitis and seizures. In very rare cases, intracranial abscesses can develop. The therapy of choice consists of complete excision of the findings, depending on the extension also in cooperation with a neurosurgeon. Recurrences are rarely observed after complete resection [44].
2.3 Ventilation and functional disorders
2.3.1 Silent sinus syndrome
The silent sinus syndrome is usually a unilateral disease of the maxillary sinus that is associated with a reduced volume, collapse of the orbital floor and resulting downward displacement of the bulb (hypoglobus) [45]. Already in 1964, Montgomery described 2 cases of enophthalmos with maxillary mucoceles [46], the term of silent sinus syndrome came up in the context of the description of a case series of 19 patients with enophthalmos and a unilateral collapse of the maxillary sinus [47]. In extremely rare cases, the ethmoid or the frontal sinus are affected [48] [49]. Worldwide, 98 cases have been described up to now [2].
Affected patients are mostly free of sinonasal complaints, however, they sometimes report about pressure of the affected side of the maxilla [50] [51]. Despite the changed visual axis due to the enophthalmos and hypoglobus, visual disorders are only rarely present because of the slowly progressing symptoms.
The most probable origin for the development of the silent sinus syndrome is an obstruction of the ostium of the affected paranasal sinus. Consecutively, negative pressure occurs as well as retracted bony walls and subsequent retention of secretion [52] [53] [54] [55] [56]. The original hypopthesis of maxillary sinus hypoplasia could be refuted by means of trials that showed computed tomographic scans from the time before the disease where a normally configured maxillary sinus was seen [54] [57] [58]. Also chronic rhinosinusitis is discussed as possible origin [56].
Endoscopy reveals a severely retracted, sometimes atelectatic uncinate process. Computed tomography reveals a seemingly hypoplastic maxillary sinus with regular retraction of all walls. The lumen of the sinus may be free or obstructed with secretion [59]. Coronary tomography may depict the orbital floor retracted in caudal direction which leads to a consecutive obstruction of the orbital content ([Fig. 6]). Magnet resonance imaging shows a hyperintense signal in the T2 weighting with homogenous imaging of the lumen [51] [59] [60] [61].
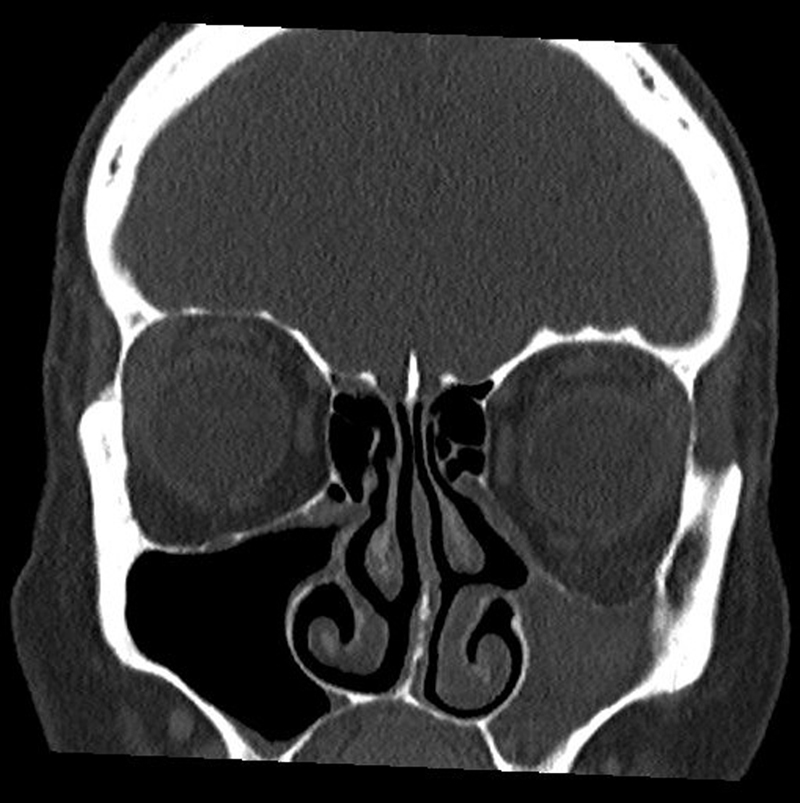
In cases of complaints, maxillary sinus fenestration type 2–3 [56] [62] [63] [64] is indicated to restore ventilation. After opening the lumen, normal to slightly hypertrophic mucosa is seen and according to the findings also mucocele-like secretion that is suctioned.
In cases of visual disorders, the correction of the orbital floor is intensively discussed. For reconstruction or lifting of the orbital floor alloplastic or autologous material may be used [55] [65]. Several case reports, however, showed regression of the enophthalmos and hypoglobus [52] [57] [65] [66] so that surgical therapy should be performed one year after restoration of the maxillary sinus ventilation at the earliest in order to allow spontaneous remission [62] [65] [67].
2.3.2 Hypersinus
The term of hypersinus describes a paranasal sinus that significantly exceeds the usual borders with a normal configuration of the bony borders and ventilation situation [68]. The extent of the affected sinus is within the borders of the cranial bone without – as in cases of pneumosinus dilatans or pneumocele – displacing it. Hence, the patients do not have any esthetic or functional impairment. [Figure 7] shows the bilateral pneumatization of the frontal sinuses. The sagittal reconstruction also depicts the frontal sinus reaching in cranial direction, however, without impairing the outline of the front.

Hypersinus does not cause any complaints so that this anatomical variation is not considered as pathology. Despite a classification established in 1987 [69], the terms of hypersinus, pneumosinus, and pneumocele are sometimes used as synonyms so that the differentiation of the hypersinus is mentioned in this context.
2.3.3 Pneumosinus dilatans
Pneumosinus dilatans is a massive extension of usually one paranasal sinus and manifests in most cases at the frontal sinus. Also the ethmoid, the sphenoid, and the maxillary sinuses may be affected by the extraordinarily strong pneumatization [56]. Protrusions of the frontal bone or intracranial, ethmoidal, and orbital extension may be found [69]. The disease is not always associated with complaints but in some cases the sense of pressure and cephalgia may result. While the affection of the frontal sinus can lead to esthetic impairment of the patients, case reports have been published describing ophthalmological complications in the context of ethmoid or sphenoid location [70] [71] [72] due to a compression of the optic nerve.
The number of cases reported worldwide amounts to 134 [73] [74]. The etiology of this disease is unknown. Possible origins are spontaneously draining mucoceles, infection with gas-forming microorganisms, genetic predisposition or fibro-osseous dysregulation as well as hormone-related dysregulation [74]. The currently most probable hypothesis is a valve mechanism of the draining pathways of the paranasal sinuses that leads to a slow and regular extension of the sinus due to the increased pressure and to spontaneously draining mucoceles. Publications of the last 20 years further indicate an association with meningiomas and arachnoid cysts [73] [75] [76] [77].
Nasal endoscopy mostly shows inconspicuous findings of the middle nasal meatus and the affected ostia if they can be assessed. Computed tomography reveals a regular protrusion of the affected sinus (mostly), generally without thinning of the bone. [Figure 8] shows a coronary CT scan of a patient with pneumosinus dilatans. The sagittal reconstruction describes the protrusion of the anterior wall of the frontal sinus as well as the altered nasofrontal angle.

A curative therapy approach is currently not available. Assuming a valve mechanism, a functional sinus surgery may be taken into consideration with dilation of the ostium and thus elimination of the stenosis. For patients with esthetic impairment because of pneumosinus dilatans frontalis, techniques of surgical modelling of the anterior wall of the frontal sinus were described by Draf et al. [78]. In cases of visual problems, the decompression of the optic nerve may be discussed in dependence of the complaints and the location of the pathologic findings. Even pneumosinus dilatans of the maxillary sinus may develop which appears primarily as outward deformity [56].
2.3.4 Pneumocele
Pneumoceles are extensions of a paranasal sinus beyond the normal borders. In contrast to pneumosinus dilatans, irregularities of the bony borders of the affected sinus with thinning and partly integrity loss are found [69] [79]. The symptoms are similar to the ones of pneumosinus dilatans. When located in the maxillary, frontal, and ethmoid sinus, a displacement of the orbital content with consecutive bulbar protrusion may result [80] [81] [82]. One case report describes temporary visual loss in the context of pneumocele of the sphenoid sinus [83] [84].
As origin for the development of pneumocele, a valve mechanism in the area of the ostium is assumed that inhibits a rapid pressure equalization between the nasal cavity and the affected sinus.
In cases of bulbar protrusion, the orbita may be decompressed by resecting the lamina papyracea. If the optic nerve is compressed in the area of the sphenoid sinus because of imbalanced pressure of the nasal cavity and the sphenoid sinus, the surgical restoration of the ventilation of the sphenoid sinus is the therapy of choice. In the above-mentioned case report, a polyp was resected that had obstructed the ostium.
2.3.5 Organized hematoma
Sinonasal organized hematomas are a rare benign disease. Repeated bleedings occur that possibly develop a mass due to a very small ostium and/or insufficient mucociliary clearance of the affected sinus. In the further course, fibrosis and neovascularization occur. Because of the expansive growth, surrounding structures may be destroyed so that imaging may be similar to malignant growth or a pathology with locally aggressive expansion of an inverted papilloma or fungal sinusitis [85] [86]. Several trials have analyzed the characteristics of the disease, however, without providing exact data regarding the incidence [85] [86] [87].
Primary complaints are often recurrent epistaxis, pains, pressure sensation in the face, and sometimes hypesthesia in the supply area of the infraorbital nerve [85] [86] [87] [88].
CT scan and MRI reveal sinuses with expansive tissue masses, frequently with extension to the ipsilateral nasal cavity [87]. Locally aggressive growth may lead to an expansion into the ethmoid sinus, orbita, pterygopalatine fossa and infratemporal fossa, cheek, and even the hard palate. Computed tomography depicts heterogenic areas with irregular spotty contrast enhancement. Calcifications may occur. MRI shows comparable contract enhancement in the T1 weighting with rather hypointense rim.
Histopathology shows bleedings with fresh and older areas, extensively dilated vessels, amyloid material with irregularly configured vessels, zones with clear neovascularization as well as hemosiderin deposits and fibroses [87].
Explorative endoscopy under general anesthesia is recommended for diagnosis and therapy. The hematoma should be removed via an endonasal access. In the context of the intervention, frequently diffuse bleedings occur. In order to prevent further encapsulation of the findings, it is recommended to create sufficiently wide accesses to the affected sinus. Recurrences are rarely described in the currently available literature [85] [86] [87] [88].
The expected endoscopic and radiological findings regarding silent sinus syndrome, organized hematoma, and pneumosinus dilatans are listed in [Table 2].
|
Disease |
Endoscopic findings |
Radiological findings |
|---|---|---|
|
Silent sinus syndrome |
Lateralization of the uncinate process |
CT scan:
|
|
Organized hematoma |
Increasing tissue in the middle nasal meatus and the nasal cavity, fibrin, granulations. Partly polypous mucosal swellings and protrusion of the lateral nasal wall |
CT scan:
MRI:
|
|
Pneumosinus dilatans |
Inconspicuous findings |
CT scan: Enlargement of the maxillary sinus beyond the natural borders without thinning of the bony walls |
2.3.6 Young syndrome
Young syndrome was first described by the urologist David Young who found that 54% of the patients with obstructive azoospermia had pulmonary defects [89]. In 1978, sinonasal complaints were included in the list of symptoms. Since then, the disease is defined as triad of obstructive azoospermia, chronic rhinosinusitis, and pulmonary ectasia or chronic bronchitis [90] [91].
With regard to the incidence of Young syndrome, no reliable data are available. Mercury exposition seems to be associated with the development of the disease. This assumption seems to be confirmed by the fact that the initially described high incidence of 1:500 males is reduced to very few cases today which might be due to the general elimination of mercury from industry and medicine [92]. According to the current stage of knowledge, a positive family history is no predisposition for the development of the disease.
Young syndrome affects young males. The initial reason why they seek medical advice is mostly infertility, only rarely chronic sinonasal or pulmonary complaints. The chronic sinonasal complaints disappear at the end of adolescence while pulmonary complaints persist [93] [94] [95].
The mucociliary clearance of affected patients is significantly longer, which, however, is not a specific diagnostic criterium [96]. Initially assumed structural deficits of the dynein arms within the cilia could not be confirmed as origin. Instead, currently an altered consistency of the nasal mucosal film is discussed that is made responsible for the patients’ complaints [91].
Differential diagnosis must exclude cystic fibrosis, primary ciliary dyskinesia, and Kartagener syndrome.
Wang et al. analyzed a cohort of 33 patients with obstructive azoospermia and described 4 patients with documented history of chronic rhinosinusitis, conspicuous imaging of the paranasal sinuses, positive family history as well as medication that may impair the mucociliary clearance. As the number of the cases described in the literature since the first description has significantly decreased and because of the definition of chronic rhinosinusitis that was inconsistently used for a long time, the existence of Young syndrome was even doubted by Arya et al. in 2009 [91].
2.3.7 Primary ciliary dyskinesia
Primary ciliary dyskinesia is a structural and functional disorder of the mobile cilia of the nasal and paranasal mucosa that leads to chronic sinonasal and pulmonary complaints. Primary ciliary dyskinesia is characterized by breathing complaints in infants, early year-round cough, and nasal obstruction. Because of the missing ciliary function, the mucosal film persists in the nose and the paranasal sinuses leading to purulent secretion in affected patients. The correct diagnosis is a real challenge because numerous diseases exist with similar symptoms. The Kartagener syndrome is a triad of chronic rhinosinusitis, bronchial ectasia, and the presence of situs inversus as consequence of ciliary dyskinesia [97].
The reason is a genetic disorder that leads to a disturbed ultrastructure of the cilia of the nasal mucosa and thus to their functional loss. Currently, 33 genes are known that are associated with the development of primary ciliary dyskinesia; the majority of them follows an autosomal-recessive inheritance [97]. The prevalence amounts to 1:15 000 live births.
In the context of primary ciliary dyskinesia, mutations of genes that code for axonal structures lead to functionally impaired cilia. In cases of primary ciliary dyskinesia, defects may include outer dynein arm defects, inner dynein arm defects, central microtubular anomalies, radial radius defects, and outer ultrastructural anomalies. Also the sperm tail and the fimbria of the fallopian tube have mobile cilia so that infertility may occur in men and women. Anatomical anomalies are possible because the defect of the mobile cilia leads to an abnormal thoraco-abdominal development during embryogenesis. Situs inversus is seen in 50% of the cases of primary ciliary dyskinesia because the regular movement of the cilia is disturbed and visceral rotation thus occurs accidentally [97] [98] [99].
The diagnosis if made based on a combination of symptoms and the results of nasal or bronchial brush biopsy for confirmation of a disturbed ciliary ultrastructure and ciliary motility. The analysis of the nasal mucosa with high-velocity video-microscopy for assessment of the ciliary motility is very sensitive and specific.
In patients older than 5 years, nasal nitrogen monoxide measurement is sensitive and may facilitate the diagnosis. The content of the nitrogen monoxide produced by the mucosa is significantly lower in patients with primary ciliary dyskinesia compared to healthy individuals. Because of the partly similar symptoms, sweat-chloride test and also genetic tests are reasonable in order to exclude cystic fibrosis [97] [100] [101] [102].
Large randomized long-term trials regarding the therapy of primary ciliary dyskinesia are not available so that many care-related aspects are based on empirical recommendations of other pulmonary diseases with similar pathologies. Despite the attempt to find a European consensus from the experience of important specialized centers, there are enormous differences regarding the approach to treat the disease [103] [104].
Regular care in narrow intervals by a pulmonologist is required. Regular spirometry, sputum cultures, and chest x-ray controls are recommended. Especially in children, the regular ENT-specific follow-up is necessary because of recurrent otitis and resulting conductive hearing loss.
Nasal symptoms usually manifest as rhinorrhea and nasal obstruction. Polyp development in affected children is rarely observed. Prophylactic antibiotic therapy may help to reduce the infectious element of rhinosinusitis. The indication of sinus surgery should be made reluctantly because its effect is controversially discussed. There is no evidence for the benefit of intranasal steroids, however, they may be helpful in the treatment of additional allergic rhinosinusitis. Improved genetic diagnosis is the first step towards a future gene-based treatment strategy such as for example gene replacement therapy, aminoglycoside-induced translational read-through, and pharmacogenetic approaches [105].
2.4 Tumors
Tumors of the sinonasal tract and the anterior skull base may develop primarily in this region or have their origin in a remote location of the head and neck area, however, manifesting within the sinonasal tract or at the skull base. The classification of the World Health Organization of 2017 indicates another group of neoplasms. Their occurrence within the sinonasal tract and at the anterior skull base is important from the point of view of differential diagnosis. The following classification of benign and malignant entities of the sinonasal tract and the anterior skull base was made based on the classification of the WHO [106].
2.4.1 Benign tumors
Benign tumors of the sinonasal tract are classified into three main categories that will be described in the following chapters:
-
Tumors of the soft parts, nerves, and vessels
-
Bone tumors
-
Other soft part tumors
2.4.1.1 Tumors of the soft parts, nerves, and vessels
2.4.1.1.1 Mucosal papillomas
Three different variations of mucosal papillomas are described. They all have in common the development from the so-called Schneiderian membrane that lines the nasal cavity and the paranasal sinuses [107] [108] [109] [110]. The ciliated mucosa of ectodermal origin develops as an invagination of olfactory ectoderm in the 4th week of embryonic development [111]. From a pathological point of view, the difference is made between 3 sinonasal (Schneiderian) papillomas:
-
Exophytic papilloma
-
Oncocytic papilloma
-
Inverted papilloma
Exophytic papilloma
This entity is also known as fungiform of septal papilloma and represents 6–50% of all Schneiderian papillomas. In contrast to the inverted papilloma, it appears mainly in males around the age of 20–50 years. In most cases it manifests at the anterior nasal septum, sometimes also at the lateral nasal wall. Multifocal occurrence is possible, bilateral manifestations have only very rarely been described [112]. Manifestation in the sinuses is extremely rare. The exophytic papilloma imposes macroscopically as rosy to grey mass with pleated surface. The therapy of choice is its excision. Malignant degeneration has not been described.
Oncocytic papilloma
With 2–26%, this entity is the rarest appearance of Schneiderian papillomas. The gender distribution is nearly balanced, a manifestation is mostly apparent after the 5th decade of life. Oncocytic papillomas develop exclusively at the lateral nasal wall, the ethmoid or maxillary sinus [113] [114]. They are very similar to inverted papillomas so that some authors describe oncocytic papillomas as microscopic variation of inverted papillomas [111] [115] [116]. Malignant degeneration is possible – in analogy to inverted papillomas (see the following chapter).
Inverted papilloma
Inverted papillomas represent the most frequent entity of Schneiderian papillomas with 47–78%. The have a polypous, mostly lobular growth, microscopically the epithelium grows downward into the mucosal stroma. In 48%, the site of origin is the ethmoid sinus, in 28% the maxillary sinus, in 7.5% the sphenoid sinus, and in 2.5% the frontal sinus. Also manifestations at the mucosa of the nasal septum are possible. Typically, unilateral manifestation is found. Bilateral manifestation has only rarely been described [117] [118]. Secondary metachronous malignant degeneration has been observed in up to 4% of the inverted papillomas with squamous cell carcinoma as most frequent entity. In cases of recurrence of an inverted papilloma, this rate increases to up to 11% [119] [120] [121].
The incidence of inverted papillomas amounts to 0.5–1.5:100 000 people per year with an age peak between the 5th and 6th decade. Males are more frequently affected (m:f 2–5:1) [108] [118] [122].
Patients suffering from inverted papilloma report about nasal obstruction, epistaxis, and epiphora if an affection of the lacrimal drainage or the inferior nasal meatus is present. Depending on the location and invasive growth behavior, mucoceles or bulbar protrusion may develop.
Clinically, mostly an edematous, rather transparent polyposis is seen. However, the appearance is highly variable because the polypous masses may impose as inflammatory and fleshy [123].
Computed tomography is the imaging technique of choice because bone erosions indicating malignant transformation become visible. Often, hyperostosis or sclerosis of the bony borders is found at the point of origin of the inverted papilloma. Calcification within the mass may also occur [124].
The therapy of choice consists of excision including the directly surrounding mucosa. A safety margin of 1–1.5 cm should be considered [62] [125] [126]. Furthermore, the removal or thinning of the bone in the area of the point of origin is required. Recurrences occur frequently if the resection has been insufficient [127]. In cases of defensive polypectomy or local excision, recurrences have been described in up to 78% [128]. Within the last few years, more efficient accesses to all areas of the maxillary sinuses could be established due to the introduction of medial maxillectomy and its variations [64]. This technique has mostly replaced the former Caldwell-Luc access [129] [130]. A review article with position paper showed equal, if not even better outcomes for endoscopic resection of inverted papillomas. If the findings are located in the maxillary sinus, the endonasal access is preferred to the open approach with regard to the incidence of recurrences [121] [131]. If a prelacrimal access is chosen, the recurrence rate is even lower [125]. In general, long-term results are the better, the more thorough the pathologic mucosa is resected [118].
2.4.1.1.2 Leiomyomas
Leiomyomas are benign tumors with muscular differentiation. In cases of angioleiomyomas, additionally a vascular differentiation is present.
In the head and neck region, they are found extremely rarely and represent not even 1% of all leiomyomas occurring [132]. In most cases, the patients are adults without preference of one gender. Most leiomyomas occurring in the sinonasal tract are have a vascular differentiation [106] [132] [133].
The tumor growth remains unnoticed and leads to unspecific symptoms such as pressure sensation and slowly progredient nasal obstruction. Epistaxis and pain may develop later.
Macroscopically, nasal leiomyomas present as polypoid to nodular and well delimited lesions with a whitish/brownish cutting surface. The mass is usually located below intact mucosa. Ulcerations rarely occur. Spindle-shaped tumor cells are arranged in overlapping fascicles with oval, long, cigar-shaped cell nuclei without atypia. Eosinophilic, fibrillary cytoplasm is present. In contrast to leiomyosarcoma, there are no mitotic alterations. Angioleiomyomas additionally have prominent vessels that are surrounded by muscle cells and closely connected to them [106].
Despite their rare occurrence, malignant transformation to leiomyosarcoma is possible. Hence, therapy should not be delayed. The therapy of choice is tumor resection. In cases of complete excision, the prognosis is very good and recurrences are extremely rare [134] [135] [136].
2.4.1.1.3 Hemangiomas
In 1897, the French physicians Poncet and Dor were the first who described lobular capillary hemangiomas which they called botryomycosis hominis [137]. Originally it was assumed that this disease was transferred from horses to humans, which was refuted by Hartzell some years later [138]. Today the terms of granuloma pyogenicum, capillary hemangioma, and epulis gravidarum are used as synonyms.
Mucosal hemangiomas represent about 10% of all head and neck hemangiomas and about 25% of all non-epithelial neoplasms of the sinonasal tract [106] [139] [140].
Hemangiomas have their origin in the capillary vessels when their density becomes too high and they nonetheless keep their original architecture with trunk and ramifications as well as surrounding pericytes. A reactive development of lobular capillary hemangiomas is also discussed due to the association with traumata or manipulations and hormonal changes during pregnancy [141]. An accumulation is further reported in the context of the application of the protein kinase inhibitor Vemurafenib [142].
Lobular capillary hemangiomas (granuloma pyogenicum) occur in all ages, however, an increased incidence is observed in children and adolescent males as well as females of childbearing age. Beyond the age of 40, the gender distribution is balanced.
The lesions may grow up to a size of 5 cm. Their surface is red bluish and located under intact mucosa. The tumor is soft on palpation, compressible, and sometimes appears polypous.
Histologically, lobular capillary hemangiomas show a trunk and branch-like pattern of capillary proliferation surrounded by pericytes. The single lobuli are separated by a fibromyxoid stroma. Inflammatory infiltrations mainly occur in ulcerated surfaces [141].
The primary clinical symptom is unilateral epistaxis followed by painless obstructive masses. The most frequent manifestation sites are the anterior nasal septum and the head of the inferior turbinate. A development in the paranasal sinuses is also possible as well as an affection of the outer nose.
The therapy of choice consists of tumor excision. More extended findings may be embolized before surgery in order to reduce the risk of bleeding. Pregnant patients may expect regression after birth. Multiple recurrences must be expected mainly in children after incomplete resection [143].
2.4.1.1.4 Schwannomas
Schwannomas are benign tumors originating from Schwann cells. They are also known as neurilemmoma and benign peripheral nerve sheath tumors.
25–45% of all schwannomas develop in the head and neck region. Most frequently, manifestations are found in the mentioned area along the vestibulo-cochlear nerve. Only about 4% of all schwannomas manifest in the sinonasal tract. According to Orphanet, the incidence of benign schwannomas amounts to 6:100 000, the sinonasal occurrence can be expected to be even lower. The age span of sinonasal manifestations is rather wide with 17–81 years and has its peak at the age of 50 without preference of one gender [106] [144].
The origins are found along the branches of the 5th and 9th cranial nerve as well as the autonomous neural system. The developing tumors may affect the nasal cavity and all paranasal sinuses [144] [145].
Primary symptoms are nasal obstruction, epistaxis, hyposmia, and sometimes pains as well as the development of Horner’s syndrome. Imaging techniques show an inhomogeneous mass with low density and sometimes bone arrosion. Differential diagnosis must exclude esthesio-neuroblastoma, adenoid cystic carcinoma, and squamous cell carcinoma. [Figure 9] shows a computed tomography scan of a big schwannoma of the maxillary nerve that originates in the foramen rotundum. Magnet resonance imaging shows a hyperintense, inhomogeneous signal of the mass in the T1 weighting.

Schwannomas have a globular, well delimited configuration. The tumor is solid on palpation and has a yellowish brounish, sometimes cystic surface [106]. Histologically, schwannomas are uncapsulated tumors that are composed of so-called myxid Antoni A areas rich in cells with nuclear palisades as well as myxoid Antoni B aeas that are poor in cells. The tumor cells present a fusiform image with cytoplasmatic extensions giving them an undulated to spindle-shaped appearance. Mitoses are extremely sparse, necrotic areas do not exist [106].
In exceptional cases, degeneration of a schwannoma is possible. After complete tumor resection, recurrences are very rare. Due to the very slow tumor growth, subtotal tumor resection is possible in cases of strong adherence to crucial neurovascular structures [144]. Fibers of the affected nerve running within the tumor are generally without any function so that resection does not lead to neurological deficits [146]. In cases of cervical schwannomas, intracapsular dissection is recommended in order not to destroy the neural fascicles surrounding the tumor [147]. Due to the narrow circumstances and the small access, however, it is only possible to a limited extent in cases of schwannomas of the paranasal sinuses.
2.4.1.1.5 Neurofibromas
Neurofibromas are benign peripheral nerve sheath tumors of schwann cells, perineural-like cells and intraneural fibroblasts. Also the term of fibroneuroma is used as synonym.
Sinonasal manifestations of neurofibromas are very rare and may occur in all ages. The peak of affected patients is at the age of 50. For patients suffering from neurofibromatosis type 1, it is at the age of 35 [148]. The prevalence of neurofibromatosis amounts to 21:100 000. Neurofibromatosis is responsible for about 10% of sinonasal neurofibromas.
They are most frequently located at the nasal entrance and the maxillary sinus with predominantly unilateral manifestation. Affected patients complain about nasal obstruction, epistaxis, and pains of the affected areas [148] [149].
Neurofibromas have a shiny, fusiform, and sometimes polypoid surface and are solid on palpation [148] [149].
Also neurofibromas are unencapsulated tumors that are closely associated with neural branches. Modified schwann cells, intraneural fibroblasts, and perineural hybrid cells with coarse collagen strains as well as mast cells in a mucopolysaccharid-rich stroma determine the histopathological picture. Oval to spindle-shaped cells with undulated, pointed nuclei with thin cytoplastmatic processes extending to the stroma are present.
In cases of complete tumor resection, the prognosis is very good. Recurrences occur in 5% especially when the tumor is resected incompletely. Malignant transformation is extremely rare [150].
2.4.1.1.6 Meningiomas
Meningiomas are benign tumors of meningothelial origin. Sinonasal menigniomas originate from extracranial, disseminated arachnoid cells within nerve sheaths or vessels.
Sinonasal meningiomas are extremely rare and responsible for less than 0.1% of all primary sinonasal neoplasms, 2% of all meningiomas, and 24% of all extracranial meningiomas. The difference must be made to intracranial findings with extracranial extension to the sinonasal tract.
Patients of every age are affected and there is no predilection of the female gender – in contrast to intracranial meningiomas. The average of disease onset is 48 years with an age range of 13–88 years.
Manifestations are often found in the nasal cavity as well as the paranasal sinuses. Manifestations in only one of the two locations are even rarer. Interestingly, most tumors of this kind are located on the left side [151] [152] [153].
Patients frequently present with endonasal, polyp-like masses, nasal obstruction, epistaxis, sinonasal complaints, pains, cephalgia, exophthalmos, periorbital edema, or visual disorders [106].
Bone infiltrations and mucosal ulcerations are possible. The cutting surface of the tumor has a grey-whitish, brownish, or reddish color. Calcifications and bone fragments can frequently be identified.
Microscopically, extracranial meningiomas may have very different appearances. Often they reveal meningotheliomatous growth with indistinct cell borders. Intranuclear pseudo-inclusions and psammoma bodies are frequently observed.
Out of the 15 histological types of meningiomas, meningothelial, transitional, metaplastic, and psammomatous tumors develop in the sinonasal tract. Most tumors may be classified as WHO grade I. Sinonasal manifestations of meningiomas grade II (atypical meningiomas growing rapidly) or grade III (anaplastic meningiomas with infiltrative growth) are extremely rare [154] [155].
Differential diagnosis must exclude carcinomas, melanomas, or aggressive psammomatous ossifying fibromas.
The complete surgical excision is the therapy of choice even if recurrence rates of up to 30% are reported. In cases of meningiomas that cannot be completely resected radiotherapy may be applied to inhibit their growth [156] [157]. The overall prognosis of sinonasal meningiomas is favorable. Metastasis and malignant transformation are not described [106].
2.4.1.2 Bone tumors
2.4.1.2.1 Osteomas and Gardner’s syndrome
Osteomas are benign, slowly growing tumors of the cranial bone that manifest frequently in the paranasal sinuses and the skull base. Most frequently, the bone proliferations are found in the frontal sinus (70–80%), the ethmoid sinus (20–25%), the maxillary sinus (5%), and extremely rarely the sphenoid sinus. The incidence of osteomas in the paranasal sinuses amounts to about 3% [118] [158] [159]. Thus, according to the definition, osteomas are no rare disease of the paranasal sinuses. However, they may be a symptom in the context of Gardner’s syndrome so that they will be discussed also in the present article.
The etiology of the tumors is not fully clarified. Current theories assume embryonic malformations, traumatic or inflammatory triggers, genetic predisposition, and disorders of the calcium metabolism as origin of the disease [118] [158] [160].
Only about 10% of all osteomas of the paranasal sinuses become symptomatic. Complaints of affected patients are often associated with obstructions of the drainage of the paranasal sinuses, i. e. recurrent acute sinusitis and also chronic sinonasal complaints. Pressure sensation, facial pains, and rhinorrhea are classic symptoms. When neighboring structures of the paranasal sinuses are involved, the orbita or the optic nerve may be compressed, in cases of intracranial involvement, pneumatocephalon may result [158] [161].
Computed tomography shows hyperdense, homogenous, well delimited areas. Magnet resonance imaging may help excluding ossified fibromas or fibrous dysplasia in the context of differential diagnosis [118].
According to the current consensus, asymptomatic osteomas should be treated by means of “wait and scan” strategy [162] [163] [164]. Regular CT controls every 2 years reveal information about the growth rate of the tumor [165]. In cases of symptomatic osteomas, the possibly complete resection is the method of choice. Depending on the location, endonasal or open surgical procedures are applied.
Gardner’s syndrome
With an incidence of 1:8000, Gardner’s syndrome is considered as rare disease [166]. In the USA, the prevalence currently amounts to 1:1 000 000 people. Patients suffering from Gardner’s syndrome often have (multiple) osteomas, soft part tumors, and intestinal polyposis (especially in the colon). Gardner’s syndrome is inherited autosomal-dominantly.
A genetic correlation with the development of Gardner’s syndrome was identified in a mutation of the gene for adenomatous polyposis coli (APC) located on chromosome 5. This tumor suppressor gene is responsible for the production of the APC protein regulating the cell growth in the cell cycle [167] [168] [169].
Regular colonoscopy is obligatory for affected patients. If APC gene mutation is confirmed, the development of colon cancer is considered as sure as of an age of 40 years so that colectomy is recommended if 20 or more colon polyps are found [166]. Hence, differential diagnosis should always exclude Gardner’s syndrome if multiple osteomas are present.
2.4.1.2.2 Fibrous dysplasia
In cases of fibrous dysplasia, mesenchymal bone development disorder is found that is caused by postzygotic somatically activating mutations. This leads to an activation of the adenylyl cyclase and increased cyclic AMP that affects the subsequent signaling pathways and causes the substitution of healthy bone by fibrous tissue and abnormally structured bone [170]. The incidences amounts to 1:4000–10 0000 [171] [172].
Depending on the location of the bone disease centers, the patients report about cephalgia and pressure sensation. If located at the tabula externa of the cranial bone, deformities with respective esthetic impairment become visible. Manifestations at the ostia or key points of the sinus drainage may lead to sinonasal complaints that can mask the basic disease for a long time.
Because of the rare occurrence of the disease, only few data exist on the treatment of fibrous dysplasia. Causal therapy currently does not exist. Pain reduction was reported after the application of bisphosphonates [173] [174] [175]. Suppression of the osteoblastoma activity, however, could not be found [176]. In cases of asymptomatic course, wait-and-see is the preferred strategy. Functional impairment and complaints such as cephalgia justify surgical procedure depending on the location of the manifestation. Prophylactic surgical therapy is not recommended, instead regular imaging controls should be performed according to available data [177] [178].
2.4.1.2.3 McCune-Albright syndrome
McCune-Albright syndrome was first described in 1936 as a triad of fibrous dysplasia, café-au-lait spots of the skin and precocious puberty [179]. The clearly variable phenotype that is known today makes McCune Albright syndrome an interdisciplinary challenge.
The reason for the development of the syndrome is a mutation of the GNAS1 gene (guanine nucleotide binding protein alpha stimulating activity polypeptide 1) which is located on chromosome 20.
The rareness of this disease leads to an insufficient characterization of the sinonasal involvement in patients with McCune-Albright syndrome. Current knowledge about the symptoms comes mainly from case reports describing complications of the disease. The prevalence of the disease is estimated to 0.55:100 000 [2].
Patients with McCune-Albright syndrome primarily suffer from weakness of the extremities or pain sensation. Most frequently, the proximal femur is affected. Fractures of the affected bone areas are often seen in childhood with decreasing incidence in direction of adolescence [180]. Bone deformations under strain are characteristic leading to the craniofacial stigmata that are typical for the disease. Craniofacial manifestations of fibrous dysplasia show a slow growth with painless swelling that sometimes lead to a clear asymmetry of the midface ([Figs 10] and [11]). Mild courses are often diagnosed accidentally in the context of X-ray of the teeth and of computed tomography performed in cases of polytrauma [170]. Severe courses frequently lead to pain sensation, paresthesia, occlusion disorders, hearing impairment, and visual disorders [170] [178] [181] [182]. In up to 50%, affected patients have impairment of the thyroid function, mostly hyperthyroidism. In about 15% of the patients, GNAS leads to an increase of prolactin and GH in the pituitary gland. The latter leads to characteristic craniofacial changes that are obvious in most patients [183].


DeKlotz et al. could show craniofacial alterations in 112 patients of a cohort of 130 patients with McCune-Albright syndrome [182]. 33% of them report about cephalgia or facial pain, nasal obstruction was found in 29% of the cases and chronic sinonasal complaints as well as hyposmia in 7%, respectively. Progress of the sinonasal involvement of fibrous dysplasia after adolescence is rare. Severe complications in the context of normal progress of the disease are rarely found ([Fig. 12]).

Such as for fibrous dysplasia, there is no causal therapy. Bisphosphonates do not show relevant therapy success [184] [185] [186]. According to the general consensus, the conservative procedure is favored, while surgical measures are applied in cases of significant symptoms or compression of vital structures [187] [188] [189] [190].
2.4.1.3 Other soft tissue tumors
In the following chapter, rare benign entities of the sinonasal tract will be discussed that have an epithelial, odontogenic, and neuroglial origin. Those are:
-
Respiratory epithelial adenomatoid hamartoma
-
Sinonasal ameloblastoma
-
Chondromesenchymal hamartoma
-
Nasal glioma
-
Cholesteatoma
2.4.1.3.1 Respiratory epithelial adenomatoid hamartoma (REAH)
Respiratory epithelial adenomatoid hamartomas have been described in 1995 for the first time [191]. In the context of this disease, tumor-like proliferation of glands in the stroma develop that is covered by multiple-row ciliated epithelium. Manifestations are found in the paranasal sinuses, the nasal cavity, and the nasopharynx. Uni- or bilateral manifestations as well as associations with chronically polypous rhinosinusitis are possible. The number of cases described worldwide fluctuates between 60 [191] [192] [193] [194] [195] [196] and about 200 [197] [198]. Because of its low incidence, the syndrome is considered as rare disease.
Especially males between 30 and 90 years of age are affected by the neoplasm [191] [195]. The symptoms usually correspond to those of chronic rhinosinusitis, i. e. pressure sensation and nasal obstruction, epistaxis and rhinorrhea, facial pain and hyposmia [191] [192] [193] [199] [200].
Most frequently, the epithelial adenomatoid hamartoma in the nasal cavity manifests at the posterior nasal septum. Frequently, both sides are affected [191] [192] [193] [194] [195] [196] [199]. Often, it is diagnosed in the context of nasal polyposis. In cases of manifestation at the olfactory fossa, clear enlargement may develop which is relevant with regard to differential diagnosis of nasal polyposis [201] [202]. An enlargement of the olfactory fossa in cases of local affection, missing contrast enhancement in the CT scan and otherwise inconspicuous paranasal sinuses, should lead to the suspicion of epithelial adenomatoid hamartoma.
Due to the initial impression, the diagnosis is often confirmed by biopsy. Regarding therapy, the complete excision should be performed. Recurrences have not been observed in the trials that are available within a period of 5 years [191] [193] [199] [203] [204].
2.4.1.3.2 Sinonasal ameloblastoma
Ameloblastomas are benign, but aggressive odontogenic tumors in most cases affect the mandible. Only about 15% occur in the maxilla. A subgroup are sinonasal ameloblastomas that probably originate from the epithelial lining of the paranasal sinuses. Locations of manifestation are the paranasal sinuses, in some cases also the involvement of the nasal cavity.
Primary symptoms are a painless mass leading to nasal obstruction and pressure sensation. In contrast to ameloblastomas of the maxilla, sinonasal variations show a solid image with partly opacification in radiography [205].
Histologically, sinonasal ameloblastomas are identical with manifestations of the oral cavity, with classic characteristics of palisaded columnar basal cells surrounding a central proliferation, similar to the radial reticulum of a developing tooth. In the paranasal sinuses, an ameloblastoma-like proliferation can be depicted under the intact mucosa. These findings are – with at the same time absent connection of the maxilla – a confirmation for a primary sinonasal origin of the tumor. In contrast to the gnathic variation, the prevalence of the sinonasal ameloblastoma is increased in males. Overall, the incidence amounts to about 0.5:100 000 000 [206] [207]. The age peak is at about 60 years [208] and thus about 15–20 years higher than the variation localized at the jaw [106].
The treatment success and thus the absence of recurrences depend on the complete surgical excision of the findings. Detailled imaging is essential in order to avoid residues. In most cases, recurrences occur within 1–2 years, however, they may also develop after a clearly longer time [205]. Descriptions of mortality caused by sinonasal ameloblastoma, metastasis or malignant transformation do not exist in the current literature.
2.4.1.3.3 Chondromesenchymal hamartoma
Chondromesenchymal hamartomas are benign, slowly growing masses with locally destructive, tumor-like growth and different mesenchymal parts. The terms of nasal chondromesenchymal hamartoma and mesenchymom are used as synonyms.
The tumor is rare and mostly occurs in children, rarely also in adolescents and adults with a slight predilection of the male gender. In the current literature currently 60 cases have been published [209].
Paranasal sinuses, nasal cavity, and orbita may be affected. Extension along the skull base and in intracranial direction are possible [209] [210].
Affected patients complain about symptoms of nasal obstruction and pressure sensation of the affected region. Because of the destructive growth with bone arrosion, imaging may lead to the impression of malignancy.
Macroscopically the solid and whitish tissue looks like cartilage. Microscopically, a lobular proliferation pattern of mature and immature hyaline cartilage is seen with various cellular and fibrous background. The cartilage and stroma parts can be penetrated with bony trabecular structures or surround bony islands [106] [211].
After complete tumor resection, the recurrence rate is very low and the prognosis is favorable.
2.4.1.3.4 Nasal glioma
Nasal gliomas (also known as heterotopic CNS tissue) are accumulations of heterotopic neuroglial tissue. Manifestations may occur in the nose and at the outer nose. In 60% the mass appears at the nasal ridge, in 30% within the nasal cavity. Even rarer, in about 10% of the cases, the tumors are diagnosed in both locations that are connected through a defect of the bone [212].
In most patients, the tumor is already present at the time of birth. About 90% of the cases are diagnosed at the age of 2 years with an equal distribution over both genders.
Clinically, a well delimited, smooth tumor is seen that is localized submucously and cannot be compressed.
In cases of findings located within the nasal cavity, nasal obstruction may occur that is the main symptom of nasal glioma – beside the esthetic impairment of findings located at the nasal ridge.
More rarely, manifestations are found in the paranasal sinuses, pharynx, nasopharynx, tongue, palate, tonsils, and within the orbita [213].
In contrast to paranasal celes, gliomas do not increase in cases of venous congestion and do not pulsate. This may be differentiated by means of the Fürstenberg test consisting of compression of the jugular vein. Additionally, CT scan and MRI reveal a soft tissue mass without intracranial part or a bony defect at the transition to the anterior cranial fossa.
Macroscopically, the tumor appears as polypoid, soft, grey-brownish mass with a size of 1–3 cm. Microscopically, the tumor is uncapsulated and composed of islands of glia tissue with different size. Inbetween, astrocytes and ribbon-like strains of vascularized connective tissue are found. The glia tissue flows into the stroma of the dermis. Mitosis is not found.
Differential diagnosis must exclude nasal encephaloceles where CNS tissue with easily identifiable neurons is found in comparison to nasal gliomas. Nonetheless, recurrences may lead to fibrous changes of the tumor that make an exact differentiation of a nasal glioma very difficult.
Complete excision of the nasal glioma is the therapy of choice. In cases of incomplete resection, recurrences may occur in about 30% of the cases. Locally aggressive behavior or a tendency of malignant transformation are not observed [212].
2.4.1.3.5 Cholesteatoma
Cholesteatomas are chronic-putrid inflammations caused scattered keratinizing squamous epithelium leading to bone destructions. Typically, these pathologies are found in the middle ear, however, nearly 30 case reports are found in the current literature describing manifestations within the nasal cavity [214] [215]. The most frequent manifestation site is the frontal sinus, followed by the maxillary sinus and the ethmoid cells. A recent report presents a manifestation in the sphenoid sinus ([Figs 13] and [14]).


The origin may be a congenital cell scattering in the context of embryogenesis or secondary scattering e. g. by surgical interventions. Also metaplasia in the context of chronic inflammations are discussed as origin [215].
The complaints depend on the location and may include cephalgia, visual disorders, pressure sensation, rhinorrhea, and failure of the cranial nerves.
The therapy of choice is the surgical and possibly complete resection that under certain circumstances is not always possible. The objective of the surgery should be a possibly wide drainage of the affected cavity in order to allow postoperative controls and if needed cleaning if the cholesteatoma could not completely be resected.
2.4.2 Malignant tumors
Malignomas of the nasal cavity and the paranasal sinus represent 0.2–0.8% of all malignant neoplasms in humans [10]. The incidence of malignomas in the nasal cavity and the paranasal sinuses amounts to 1.5:100 000 in males and less than 1:100 000 in females. Consequently, all malignant entities in this anatomic region must be considered as rare diseases.
2.4.2.1 Malignant epithelial tumors
2.4.2.1.1 Squamous cell carcinomas
2.4.2.1.1.1 Keratinizing squamous cell carcinoma
Sinonasal squamous cell carcinomas are malignant neoplasms that originate at the surface of the epithelium of the nasal cavity and the paranasal sinuses and have a differentiation of squamous epithelium.
The affection of the sinonasal tract by squamous cell carcinomas is rare. It is the location of the head and neck that is less frequently affected by this entity [216]. The age of disease onset is between the 6th and 7th decade of life with significantly more male patients (m:w=2:1) [106] [216] [217] [218].
Tobacco consumption increases the risk of the development of squamous cell carcinomas in the sinonasal tract, however significantly less than in other locations of the head and neck [219] [220] [221]. High-risk HPV infections are mostly associated with the occurrence of non-keratinizing squamous cell carcinomas. In rare cases, sinonasal papillomas can transform after malignant degeneration into keratinizing or non-keratinizing squamous epithelium [222].
The most frequently observed site of manifestation is the maxillary sinus, followed by the nasal cavity and the ethmoid sinus. Affection of the sphenoid or frontal sinus is extremely rare [106].
Initially, affected patients have unspecific complaints such as nasal obstruction, epistaxis, and rhinorrhea or sinonasal complaints. Pains about the region concerned, bulbar protrusion or diplopia and paralysis are symptoms of more extended disease. In older patients, it may manifest by a maxillary prosthesis that does no longer fit when the hard palate is infiltrated.
Macroscopically, the tumor grows exo- or endophytically with variable ulcerations, necrotic areas, and hemorrhagic parts. Microscopically, the tumor has identical characteristics as manifestations in other head and neck regions. Those are irregular nest formation and ribbon-like alignments of eosinophilic cells that show important keratinization and induce desmoplastic stroma reaction. The difference is made between well, moderately, and poorly differentiated keratinizing squamous cell carcinomas.
The therapy of choice is the tumor resection with adjuvant radiotherapy. In cases of inoperable findings, primary radiochemotherapy should be performed. The 5-year survival rate for sinonasal squamous cell carcinomas amounts to 50–60% and strongly depends on the tumor stage at the time of diagnosis. Carcinomas of the nasal cavity have a more favorable prognosis than manifestations within the paranasal sinuses because they only lead to complaints in higher tumor stages [216] [217] [218] [223] [224].
2.4.2.1.1.2 Non-keratinizing squamous cell carcinoma
Non-keratinizing squamous cell carcinomas are characterized by a distinct ribbon-like growth pattern with missing or impaired maturation.
Other terms that are used are Schneiderian carcinoma, cylinder cell carcinoma or transition carcinoma.
Non-keratinizing squamous cell carcinomas represent about 10–27% of the sinonasal squamous cell carcinomas. They affect patients in the 6th and 7th decade of life, among them clearly more male patients are found [225] [226] [227] [228].
The risk factors are similar to those of keratinizing squamous cell carcinomas of the sinonasal tract, however, in 30–50% of the cases, transcriptionally active high-risk HP viruses can be identified. Between 2 and 10% of the sinonasal papillomas may transform to malignant keratinizing and more rarely to non-keratinizing squamous epithelium [106] [222].
Macroscopically, the tumor shows a variable exophytic and/or inverted growth pattern with fragile structures and necrotic as well as hemorrhagic areas. Microscopically, the tumors grow as extending foci or anastomosing ribbon-like alignments of cells in the submucosa with a lining of smooth stroma. Papillary properties can be identified within or at the surface of the tumor [106].
Similar to keratinizing sinonasal squamous cell carcinoma the endonasal or open tumor resection should be performed followed by adjuvant radiotherapy, alternatively to primary radiochemotherapy for non-resectable findings. The overall 5-year survival rate of non-keratinizing squamous cell carcinomas amounts to about 60%. HPV-associated carcinomas have better survival chances even if the prognostic significance is not so strongly supported as for tumors of the oropharynx [226] [229]. Lymph node metastases are present at the time of first diagnosis in 3.3 to 26% of the cases [230] [231].
2.4.2.1.1.3 Spindle cell/sarcomatoid squamous cell carcinomas
Spindle cell squamous cell carcinomas or sarcomatoid carcinomas of the sinonasal tract are special forms of squamous cell carcinomas that are defined by the presence of predominantly malignant spindle cells and/or pleomorphic cells.
This special type manifests mainly in older male patients. The tumor is extremely rare in the sinonasal tract and represents less than 5% of all squamous cell carcinomas in this region [226] [232] [233] [234].
The development of spindle cell squamous cell carcinomas is closely associated with tobacco consumption and exposition to radiation. In the few cases that are known up to now, no HPV infection could be identified [226] [235] [236].
Symptoms of affected patients are initially unspecific and mostly express as nasal obstruction and epistaxis. In higher tumor stages, facial swellings and diplopia as well as pains of the affected areas may occur [106].
Spindle cell squamous cell carcinomas grow as polypous masses with ulcerating surface and are similar to the macroscopic appearance of more frequently occurring laryngeal findings. They originate from squamous epithelium and show variable differentiation with epithelial mesenchymal transition. The tumors may contain residues of dysplastic squamous epithelium and often reveal areas with transition to malignant spindle or pleomorphic tumor cells [106].
With regard to the prognosis and predictive factors in cases of sinonasal manifestation, no exact data are available because of the extremely low number of cases.
2.4.2.1.2 Lymphoepithelial carcinoma
Lymphoepithelial carcinomas are poorly differentiated squamous cell carcinomas or histologically undifferentiated carcinomas. A prominent lymphoplasmatic infiltration that is similar to nasopharyngeal carcinomas is typical for the microscopic appearance of the tumor.
This entity is very rare and occurs – similar to nasopharyngeal carcinomas – mostly in Southeast Asian countries. The age peak is between 50 and 70 years, preferably in males (m:w=3:1). Etiology shows an association of lymphoepithelial carcinomas with Epstein-Barr visus [10] [237].
Manifestations can often be found in the nasal cavity, the paranasal sinuses are less frequently affected. Infiltrative growth into the palate, the orbita or the skull base are possible.
Symptoms reported by affected patients are pressure sensation, nasal obstruction, epistaxis, and in cases of infiltration of the orbita a bulbar protrusion. Neurological deficits may occur in the context of intracranial infiltration [237] [238]. Lymph node or distant metastasis is possible. Thorough endoscopy and biopsy of the nasopharynx should be performed in order to exclude loco-regional extension of a nasopharyngeal carcinoma.
Due to the low number of cases, there is no standard therapy. Because of the high radiosensitivity, most cases undergo loco-regional radiotherapy that is very effective even when cervical lymph node metastases are present. Radiochemotherapy followed by salvage surgery is possible in cases of extended findings [239]. However, it is difficult to assess the data situation because of the low number of cases.
2.4.2.1.3 Sinonasal undifferentiated carcinoma (SNUC)
Sinonasal undifferentiated carcinomas were first described in 1986 by Frierson et al. [240]. It is a highly aggressive carcinoma which show a locally extensive growth. Pleomorphic tumor cells and numerous tumor necrosis area are characteristic for the appearance. It is a high-grade epithelial neoplasm with invisible histogenesis with or without neuroendocrine differentiation. An exact delimitation with regard to lymphoepithelial carcinoma and olfactory neuroblastoma is decisive [10] [240].
The incidence is very low with about 100 cases described in the literature. The age range is between 30 and 90 years with a higher incidence in males (m:w=2–3:1) [237] [241].
There is no association with the Epstein-Barr virus. Some cases occurred after previous radiotherapy in the context of nasopharyngeal carcinoma [237].
SNUC mostly manifests in the nasal cavity, the maxillary and ethmoid sinuses with frequent infiltration of neighboring structures. In up to 50% of the cases, patients already show infiltrations of the dura and 30% infiltrations of the orbita at the time of confirmed diagnosis [242] [243]. In a trial that analyzed the percentage of the T stage in relation to the examined carcinoma histologies, 69% of the patients already had stage T4 of SNUC [244].
Similar to lymphoepithelial carcinomas, affected patients initially suffer from nasal obstruction, epistaxis, later bulbar protrusion, periorbital swellings as well as facial pains and in cases of intracranial infiltration failures of the cranial nerves.
SNUC shows a high rate of local recurrences and distant metastasis with frequent vascular and neural infiltration [245] [246]. According to a recent trial analyzing 318 patients with SNUC in the USA, the 5-year survival rate amounts to 34.9% and the 10-year survival rate to 31.3% [247]. In most cases, the poor prognosis is caused by the already high tumor stage at the time of diagnosis and the resulting inoperability. Another reason is the fact that the majority of the published case series contains less than 20 patients so that no standardized therapy approach or a guideline are available regarding the therapy of SNUC [242].
Considering the current literature, there is a consensus that multimodal aggressive therapy should be performed consisting of surgery, radio- and chemotherapy. This approach was confirmed by 2 large SEER (Surveillance, Epidemiology, and End Results program) database trials [247] [248]. The tumor resection followed by adjuvant radiochemotherapy or primary radiochemotherapy are possible therapeutic approaches depending on the resectability of the tumor. Approaches of targeted therapy with lapatinib, which suppressed the human epidermal growth factor receptor 2 (HER2) signaling pathways in vitro as well as in vivo, showed first promising results that allow prospects to more effective therapies [249].
2.4.2.1.4 Adenocarcinoma
2.4.2.1.4.1 Adenocarcinoma of the intestinal type
The morphological structure of sinonasal adenocarcinomas of the intestinal type is similar to the one of adenocarcinomas of the gut.
The occurrence is very rare and is estimated to amount to an incidence of less than 1:1 000 000 people per year. However, the incidence varies significantly and increases regarding the prevalence by factor 500 in people working in wood and leather processing industries. The carcinoma is thus a recognized occupational disease (BK4203). Men are affected 3–4 time more frequently than women which is probably due to the employment in the professions. The age of disease onset is mostly between the 6th and 7th decade of life [106].
Manifestations are often near the lateral nasal wall and the middle turbinate. According to estimations, 40% of the tumors develop in the ethmoid sinus, 28% in the nasal cavity, and 23% in the maxillary sinus [250] [251].
Symptoms are unilateral nasal obstruction, epistaxis, and rhinorrhea. More rarely, pains, facial swelling and diplopia, or bulbar protrusion may occur. Destruction of the surrounding bone and infiltration into neighboring regions are frequently observed [106] [251].
Macroscopically, a polypoid, partly papillary and nodular mass with fragile consistency and ulcerating, hemorrhagic as well as rarely gelatinous or mucous parts are seen [106] [250] [251]. Microscopically, exophytic, papillary and tubular or mucinous growth consisting mainly of signet ring cells. The grade of differentiation is highly variable. In 6–10%, KRAS mutation can be found. BRAF mutation occurs clearly more rarely with less than 10% [252] [253] [254] [255].
Because of the poor radiation sensitivity of the tumors, radical tumor resection with free resection margins is the therapy of choice. Due to the development of the endoscopic technique, the transnasal resection is possible in some cases also with orbital and intracranial infiltration [256], sometimes with consideration of complex reconstructive measures [257]. Low-grade adenocarcinomas of the intestinal type with low tumor stages can be treated by radical tumor resection. In all other stages and entities, adjuvant radiotherapy should be performed [258]. Intensity modulated photon irradiation may be discussed as part of the therapy strategy [259]. Up to now, no phase-III trials are available with regard to chemotherapy so that current outcome reports are mainly based on case reports and small retrospective case series. In the context of a prospective phase-II trial, primary complete remission could be achieved in 16% of the patients treated with neoadjuvant chemotherapy with PFL (cisplatin, 5-fluorouracil, and leucovorin) [260]. In these cases, the 3-year survival rate amounted to 100% compared to 43% in the other patients.
Low-grade papillary adenocarcinomas have the best 3-year survival rate with<80 and<60% after 5 years. Grade 2 and 3 tumors have a 3-year survival rate of 54 and 35%, respectively. Mucinous tumors with alveolar growth have similar survival rates like papillary tumors of grade 2. The most aggressive growth is found in signet ring adenocarcinomas. In cases of infiltration of the orbita, the skin, the ethmoid or sphenoid sinus as well as the skull base, the prognosis is significantly poorer. Lymph node metastases can be found in 8% and distant metastases in 13% of the cases at the time of diagnoses [106] [251].
2.4.2.1.4.2 Adenocarcinoma of the non-intestinal type
Sinonasal adenocarcinomas of the non-intestinal type do not show characteristics of salivary gland carcinomas and do not have an intestinal phenotype. The mentioned tumor category is heterogenic. Nonetheless, it has specific entities that are singular (e. g. the renal cell-like carcinoma) [106].
Sinonasal low-grade non-intestinal adenocarcinomas occur very rarely, they do not show a predilection of a specific gender and may be diagnosed within an age range of 9–89 years. The mean age at the time of diagnosis is within the 6th decade of life. High-grade non-intestinal adenocarcinomas are rare, affect male patients more frequently, also have a broad age span with a peak within the 6th decade of life. Neither for low-grade nor for high-grade non-intestinal adenocarcinomas of the sinonasal tract, the etiology is known.
Most frequently, these tumors manifest in the nasal cavity (64%), followed by the ethmoid sinus (20%). About half of all high-grade non-intestinal adenocarcinomas already have an advanced stage at the time of diagnoses with infiltration of the nasal cavity and the paranasal sinuses [261] [262] [263] [264].
Symptoms of the low-grade variation are frequently unspecific and comprise nasal obstruction, epistaxis, and pain sensation. High-grade tumors frequently cause deformities of the facial skin and bulbar protrusion when the orbita is infiltrated. Morphologically, the imaging of low-grade non-intestinal adenocarcinomas as solid mass filling the nasal cavity or the paranasal sinuses whereas high-grade non-intestinal adenocarcinomas have a clearly more destructive growth with osseous infiltration and invasion of the surrounding anatomical areas [106].
Macroscopically, low-grade tumors impose as red, polypoid or raspberry-like solid structures. Histologically, papillary and/or tubular characteristics with complex growth patterns. A single layer of uniform, mucinous, cuboid to columnar epithelial cells surrounds the tumors. High-grade tumors show significantly more histological diversity. Solid growth with occasionally interspersed glandular structures as well as singular mucocytes are typical. Some variations show nest-like distribution patterns with infiltrative behavior. Numerous mitosis types and necrotic areas are found [106].
The therapy of choice consists of radical tumor resection. Despite the sparse data situation due to few described cases, surgical therapy alone – tumor-free resection margins provided – seems to be decisive for the prognosis [265]. A recently performed National Cancer Database analysis did not reveal any influence of radiotherapy on the survival of patients with low-grade adenocarcinoma [266]. Patients with high-grade adenocarcinomas show a clearly better survival rate in case of multimodal therapy including radical tumor resection and adjuvant radiotherapy. Due to the low number of cases, the role of systemic therapy could not be definitely clarified [256].
Recurrences are found in 25% of the low-grade variation and only about 6% of the affected patients die of their disease. The prognosis for patients with high-grade non-intestinal adenocarcinomas is significantly poorer. Most patients die within 5 years after diagnosis of the disease. Local and distant metastasis occasionally develops [261] [262] [263].
In the few cases reported about renal cell-like carcinoma, neither recurrences nor metastasis were found [267].
2.4.2.1.5 Salivary gland like carcinomas
Salivary gland neoplasms in the nose or the paranasal sinuses are extremely rare. The majority of the cases consists of malignant entities. An analysis of Daniel K. Heffner shows the following distribution of the entities in a total of 311 patients with sinonasal salivary gland like tumors [268].
The following list consists of the malignant sinonasal entities:
-
Adenoidcystic carcinoma
-
Acinar cell carcinoma
-
Mucoepidermoid carcinoma
-
Epithelial-myoepithelial carcinoma
2.4.2.1.5.1 Adenoidcystic carcinomas (ACC)
Adenoidcystic carcinomas mainly occur in the major and minor salivary glands. However, manifestation may be found in all areas where secretory glands are localized (breast, cervix, colon, prostate, nasal cavity, and paranasal sinuses). It is the most frequent malignoma of the sinonasal tract with an age span of 11–92 years. Sinonasal manifestations are found in the maxillary sinus (60%) and the nasal cavity (25%). Adenoidcystic carcinomas represent 5% of all sinonasal malignant neoplasms with an incidence of 25% referring of all salivary gland carcinomas [269] [270] [271] [272] [273]. It grows slowly and infiltrates surrounding structures along the involved cranial nerves. Hematogenic metastasis is frequently observed and may occur even years after first diagnosis. Due to the late occurrence of symptoms, affected patients have high tumor stages which leads to a corresponding morbidity when the tumor is resected or primary radiotherapy is performed. Due to the low radiation sensitivity, doses of>80 Gy are required which leads to collateral damage of neighboring structures (orbita, optic nerve, cerebrum) [274] [275] [276]. The therapy of choice consists of radical tumor resection and postoperative radiotherapy for R1/R2 resection in cases of perinodal infiltration and advanced tumor stages (T3/T4), even if some trials could confirm a better outcome in all tumor stages (T1–4) [274] [277] [278] [279] [280].
Due to the high tumor stages at the time of first diagnoses and the frequently occurring late recurrences, the prognosis of patients with adenoidcystic carcinomas is poor. The 5-year survival rate amounts to 38–64% after conventional irradiation. With application of proton radiation, the local tumor control reaches 50–70% and even up to 93% in cases of neutron radiation [271] [274] [275] [281] [282] [283] [284] [285].
2.4.2.1.5.2 Acinar cell carcinoma
Acinar cell carcinomas are rare malignomas of the salivary glands that are mainly found in the parotid gland. After tumor resection with or without postoperative radiotherapy, the 20-year survival rates amount to nearly 90% [286]. Manifestation in the maxillary or ethmoid sinus, at the turbinate or the nasal septum is extremely rare. The current English literature describes 19 cases, their follow-up reaches from 1 to 22 years. 5-year and 10-year survival rates amount to more than 90% [287] [288] [289] [290].
2.4.2.1.5.3 Mucoepidermoid carcinoma
Sinonasal mucoepidermoid carcinomas are malignomas of the minor salivary glands that manifest in the oral cavity and the oropharynx and have a 5-year survival rate of 87% at these locations [291]. Lymph node metastases occur in 15% of the cases.
Sinonasal mucoepidermoid carcinomas represent about 1.5% of all sinonasal malignomas and thus they are very rare. Most frequent locations are the maxillary sinus and the nasal cavity [292] where high-grade mucoepidermoid carcinomas may be diagnosed in 46%. Symptoms of affected patients are nasal obstruction, chronic sinonasal complaints, facial pain, epistaxis, and cephalgia. Small monocentric trials show a 5-year survival rate of 35.9% to 44.1% in sinonasal manifestation [204] [293]. Tumor resection with adjuvant radiation is the therapy of choice, however, data regarding the effectiveness are not available due to the low number of cases [292].
2.4.2.1.5.4 Epithelial-myoepithelial carcinoma
Epithelial-myoepithelial carcinomas represent an extremely rare malignant entity which predominantly manifests in the major salivary glands and makes up about 1% of all salivary gland malignomas. Even rarer is the manifestation in the nasal cavity or the paranasal sinuses, only few case reports exist here [295]. The largest cohort trial includes 468 patient with epithelial-myoepithelial carcinomas. 18 of these patients had manifestations in the sinonasal area [296]. 80% of the patients were older than 50 year. Female patients were affected more frequently (f:m=1.5–6:1) [296]. Due to the low number of cases, the description of the pathophysiology and the therapeutic strategies is not systematic because a high percentage of the relevant literature consists of case reports.
The term of epithelial-myoepithelial carcinomas is a histopathological term describing the proliferation of tubular structures with two-layer cell lining. The inner layer consist of cubic or low-cylindrical duct cells, the outer layer consists of bright epitheloid cells. The tumor stroma may be hyalinized [297].
According to the location, patients present with symptoms such as epistaxis, pressure sensation and pains, swellings, and rhinorrhea. Endoscopy frequently reveals hemorrhagic masses. Computed tomography shows a heterogenic contrast enhancing soft part structure win the affected paranasal sinuses. Osseous destructions of neighboring structures are not typical, however, they may occur [295].
Current reports show only low glucose uptake in the PET/CT which is due to the low-grade potential of malignancy and makes the significance of preoperative imaging difficult with regard to the diagnosis [298] [299].
The therapy of choice is the generous surgical excision. Due to the inhomogeneous biological behavior and response, the effectiveness of adjuvant radiotherapy is unknown [300].
Lymph node metastases are rare and occur in less than 5% of the cases. The 5- and 10-year survival rates amount to 72.7 and 59.5%, respectively [296].
2.4.2.2 Neuroendocrine neoplasms
The term of neuroendocrine neoplasms comprises several entities. Beside the neuroendocrine carcinoid and the classic neuroendocrine carcinomas, also esthesioneuroblastomas (see 2.4.2.6.2) as well as sinonasal and undifferentiated carcinomas (SNUC, see 2.4.2.1.3) belong to the neoplasms with neuroendocrine differentiation [301].
Neuroendocrine neoplasms express neuroendocrine markers such as synaptophysin and chromogranin A. Large membrane-bound (hormone-containing) vesicles are characteristic. Epithelial neuroendocrine carcinomas (well, moderately, or poorly differentiated carcinoid) develop from cells of the diffuse neuroendocrine cell system, that express cytokeratins and are located in the mucosa. Blastomas of the olfactory nerve and paragangliomas are of neuroectodermal origin and develop from the olfactory membrane or head and neck paraganglia [302]. According to the WHO classification, neuroendocrine tumors are considered as grade I neoplasms (formerly known as low-grade neuroendocrine tumors or typical carcinoid), grade II neoplasms (formerly called intermediary neuroendocrine tumor or atypical carcinoid), and grade III neoplasms, small-cell carcinoma (formerly called high-grade neuroendocrine tumor or small-cell carcinoma) as well as grade III neoplasms, large-cell carcinoma (formerly known as high-grade neuroendocrine tumor or large-cell neuroendocrine carcinoma) [303].
Neuroendocrine carcinoid (typical carcinoid, grade I neoplasm)
More than 90% of neuroendocrine carcinoids manifest in the larynx (supraglottic). Sinonasal manifestations occur more rarely and lead to well delimited, submucous and often polypoid tumors [304]. Especially poorly differentiated neuroendocrine carcinomas are found in the nasal cavity, the paranasal sinuses, or at the skull base. The 5-year survival rate amounts to 5–20% [106].
The symptoms depend of the manifestation sites. In cases of excessive hormone production, carcinoid syndrome is possible, especially in cases of hepatic metastasis, and it occurs in about 10% of all patients with carcinoids. The typical triad comprises attacks of redness of the face and the trunk, diarrhea, and cardiac involvement [305].
Neuroendocrine carcinomas
Sinonasal neuroendocrine carcinomas are high-grade carcinomas with morphological and immunohistochemical characteristics of neuroendocrine differentiation [106]. They are subdivided into small-cell neuroendocrine carcinomas and large-cell neuroendocrine carcinomas and represent about 3% of all sinonasal tumors. They develop more frequently in males of middle and higher ages (mean age of 49–65 years). In rare cases, an association with high-risk human papilloma viruses is reported.
Primary sinonasal manifestation sites are the ethmoid sinus, the nasal cavity, followed by the maxillary and sphenoid sinus.
Symptoms of manifestation are mostly unspecific (nasal obstruction, rhinorrhea, and chronic sinonasal complaints). Many patients only consult their doctor with higher tumor stages.
Endoscopically, large, hemorrhagic tumor masses are found with necrotic parts. Morphologically, imaging shows osseous destructions and infiltrations of neighboring anatomical regions.
With regard to the histopathology, sinonasal neuroendocrine carcinomas are identical to neuroendocrine carcinomas occurring in the lung and other head and neck locations. They have a highly infiltrative growth behavior with frequent perineural and lymphovascular infiltration. Sinonasal neuroendocrine small-cell and large-cell carcinomas have a cytokeratin expression which allows differentiation for example with regard to olfactory neuroblastomas [303].
If possible, therapy should aim at tumor resection followed by adjuvant radiochemotherapy. Especially in cases of large-cell neuroendocrine carcinomas, neoadjuvant radiochemotherapy followed by tumor resection seems to achieve the best outcome [301].
The 5-year survival rate amounts to 50–65%. Location in the sphenoid sinus has a more favorable prognosis (~80%) compared to maxillary or ethmoid manifestations (~33%). Despite the limited data situation due to the low number of cases, the prognosis for large-cell neuroendocrine carcinomas seems to be more favorable than for the small-cell variant [106].
2.4.2.3 Malignant soft tissue tumors (sarcomas)
2.4.2.3.1 Fibrosarcoma
Fibrosarcomas are malignant spindle-cell tumors with fascicular architecture and variable collagen matrix production showing a fibroblastic/myofibroblastic differentiation. Furthermore, fibrosarcomas have low mitosis rates and rare nuclear pleomorphisms or anaplastic properties.
Manifestation sites are mostly located in the extremities, only 1% of fibrosarcomas are found in the head and neck. They are responsible for less than 3% of all non-epithelial malignomas of the head and neck, however, they are the second most frequent entity of head and neck sarcomas [106] [306].
Symptoms are similar to those of other neoplasms of the nasal cavity and the paranasal sinuses. Unspecific complaints such as nasal obstruction and epistaxis, pressure sensation, pains, and swellings may occur.
Fibrosarcomas have a high risk for local recurrences but only a low risk for distant metastasis [140] [307]. Despite the low number of cases, a generous tumor resection is recommended because small safety margins significantly increase the risk of local recurrence [140]. A database analysis of the US National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) investigated the course of 51 patients with sinonasal fibrosarcomas over a period from 1973 to 2012 [306]. The mean age amounted to 54.5 years without a predilection of one gender. Interestingly, 83.7% of the patients had bright skin and only 8.2% were dark-skinned. The most frequent locations were the maxillary sinus with 54.9% followed by the nasal cavity (23.5%). The most frequent histological type was the moderately differentiated (59.5%) followed by the well-differentiated fibrosarcoma (16.2%). The follow-up revealed local recurrence in 28.2% of the patients, in 64.1% of the cases regional metastasis developed and 7.7% had distant metastasis.
The most frequently applied therapy modality was tumor resection alone (61.2%) followed by tumor resection with adjuvant radiotherapy (32.7%). Also local recurrences were treated primarily by tumor re-resection (71.4%) followed by tumor resection with adjuvant radiotherapy (7.1%). The 5-year survival rate amounted to 71.7% in all cases with follow-up. Recent trials show clear advantages for patients who undergo adjuvant radiotherapy [307] [308].
2.4.2.3.2 Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma
The undifferentiated pleomorphic sarcoma (also known as malignant fibrous histiocytoma) is a high-grade soft part sarcoma without differentiation line. It develops in adults, sinonasal or skull base manifestation is very rare even if it ranks third after rhabdomyosarcoma and fibromyosarcoma as sarcoma in this location [106].
Previous radiotherapies seem to be responsible for the development of undifferentiated pleomorphic sacromas [309] [310].
In particular unspecific symptoms such as painless swellings, nasal obstruction, bulbar protrusion and diplopia as well as epistaxis may occur. Rather rarely, regional or distant metastasis is observed [106].
Endoscopically, a lobular grey-whitish, partly fleshy mass with hemorrhagic parts is found. Most findings seem to be circumscribed. Microscopically, the malignomas present spindle and pleomorphic cells in a variably collagenized extracellular matrix, pleomorphism, atypical mitoses, necrotic areas, histocytic and foam cells are often found. Many tumor cells have characteristics of fibroblasts, myofibroblasts, or histiocytes.
The undifferentiated pleomorphic sarcoma is diagnosed by excluding mucosal melanomas, carcinomas, lymphomas, and other sarcomas.
Despite the poor data situation and independently from the situation of the margins, tumor resection seems to be essential. Radiotherapy increases the chance of local tumor control [106] [310]. The 5-year survival rate amounts to 60–70%.
In their trial, Gerrand et al. could show for sarcomas of the extremities different outcomes depending on the histological subtype, the application of radiotherapy, the local anatomy, and unplanned excision prior to admission in a specific center [311]. They postulated that patients should be transferred to specialized diagnostic and therapeutic center with multidisciplinary sarcoma specialists if soft part sarcoma is suspected [310].
2.4.2.3.3 Leiomyosarcoma
Leiomyosarcomas are malignant tumors originating from the smooth muscles. Typical manifestations sites are the uterus or intestinal tract. Sinonasal manifestations or location at the skull base are extremely rare. Mainly adults are affected, in exceptional cases also children. Also for leiomyosarcomas, a previously performed radiotherapy plays a crucial role for the genesis [106].
Due to only unspecific symptoms, patients present to their doctors with late tumor stages which leads to poorer prognoses of sinonasal manifestation compared to other locations [312]. Clinically, a soft, mainly polypous tumor mass presents that may cause pains, nasal obstruction, and epistaxis. The lesions may also affect the craniofacial bone and cause further complaints such as diplopia, bulbar protrusion etc. depending on the infiltration.
Hematogenic metastasis into the lung, liver, other soft part areas, bones, or cerebral structures are possible. Metastases of other regions should be excluded before definitive tumor therapy.
Macroscopically, the tumor mass is polypoid. Clear delimitations of the surrounding tissue as well as findings that cannot be clearly defined are possible. Microscopically, infiltrative growth or sharp delimitations, spindle cells aligned in interconnected fascicles are characteristic. The eosinophilic cytoplasm often shows small perinuclear vacuoles [106].
Computed tomography is often unspecific and shows expansive, cystic or necrotic, heterogenic lesions within the soft tissue. MRI shows only moderate hyperintensity in the T1 and T2 weighting which makes exact diagnosis difficult [313].
Tumor resection is the method of choice, however, resection with sufficient safety margins is only possible to a limited extent due to the location and vital neighboring structures. Adjuvant chemotherapy and/or radiotherapy are applied in patients with locally advanced growth, recurrences, and metastases [312] [314]. About one third of the patients with sinonasal manifestation either dies from distant metastasis or local recurrences that grow into vital neighboring structures [106].
2.4.2.3.4 Rhabdomyosarcoma
Rhabdomyosarcomas are malignant mesenchymal tumors with skeletal muscle differentiation.
The difference is made between:
-
Embryonic rhabdomyosarcomas
-
Alveolar rhabdomyosarcomas
-
Pleomorphic rhabdomyosarcomas
-
Spindle cell rhabdomyosarcomas
The terms of myosarcoma and malignant rhabdomyoma are used as synonyms for rhabdomyosarcoma.
The incidence of sinonasal rhabdomyosarcomas amounts to 0.034:100 000 with primary manifestation in the paranasal sinuses followed by the nasal cavity. It is the most frequent sinonasal sarcoma in children as well as in adults. The age peak is within the first decade of life [106] [311] [316]. Radiation-induced genesis is currently investigated.
Macroscopically, a polypoid, poorly delimited tumor formation is seen with extension to neighboring structures and fleshy, gelatinous, brownish to grey surface.
Embryonic rhabdomyosarcomas is the entity that occurs most frequently in the sinonasal tract. It has primitive to spindle-shaped cells with sparse cytoplasm and hyperchromatic nuclei as well as dispersed rhabdomyoblasts with clearly eosinophilic cytoplasm. The number of rhabdomyoblasts typically increases significantly after radiotherapy.
In adults, more frequently sinonasal alveolar rhabdomyosarcomas are found [311] which have fibrovascular septa that separate the foci from round neoplastic cell accumulations. Giant cells with multiple peripherally located cell nuclei may be present [106].
The outcome seems to be poorer for patients who received previous excision or excisional biopsy for histological assessment compared to patients who underwent directly complete tumor resection [311] [312]. Thus it seems to be particularly important to transfer patients to an interdisciplinary center specialized in sarcomas if this entity is suspected.
As therapy, tumor resection with broad safety margins should be intended. Also in cases of incomplete tumor resection, surgery seems to be a predictive factor [311]. Five-year survival rates of 40–45% are reported in cases of combination of surgery, chemotherapy, and irradiation, for patients younger than 18 years and females, this rate is slightly higher. If the skull base is infiltrated, the prognosis is much poorer [106] [311] [317].
2.4.2.3.5 Angiosarcoma
Angiosarcomas are vascular malignant neoplasms. Other terms for this entity are epitheloid hemangioendothelioma, malignant hemangioendothelioma, malignant angioendothelioma, or hemangiosarcoma.
In more than half of the cases, the tumor develops within the skin and superficial tissue layers of the head and neck. Epidemiologically, the sinonasal angiosarcoma is responsible for less than 0.1% of all head and neck malignomas and less than 1% of all sinonasal malignomas [106].
Possible, however rarely reported risk factors for the development are radiation exposure, vinyl chloride and carbon dust exposition [318] [319] [320].
Angiosarcomas are located rather in the nasal cavity and the maxillary sinus. They lead to initially unspecific complaints, predominantly epistaxis and nasal obstruction. Some patients further report about sinonasal complaints, epiphora, pains, and pressure sensation.
Often, bone infiltration is observed with angiosarcomas that may be made visible in computed tomography. In the T2 weighting of MRI, a strong signal intensity can be depicted. Preoperative angiography may be helpful to identify afferent vessels and to allow preoperative embolization [106] [321]. Lymph node and distant metastasis are unusual at first manifestation.
Macroscopically, angiosarcomas present with nodular to polypoid, soft and livid to reddish surface with ulcerations that easily bleed. Microscopically, the malignoma develops under an intact epithelial layer with neoplastic vessel formation that extend into the soft tissue and neighboring bones. Hemorrhagic and necrotic areas are accompanying symptoms. Within the neoplasm, capillary, cavernous, and rudimentary vessels are found that are filled with erythrocytes and lined with increased, atypical spindle-like or epitheloid endothelial cells. Grading is not performed for angiosarcomas [106] [321].
Due to the very low incidence, there is no standard therapy for sinonasal angiosarcomas. In most cases, tumor resection followed by radiotherapy is described. Other therapeutic approaches that have been described for single cases are chemotherapy, gamma-knife therapy and the application of interleukins [321] [322] [323] [324]. With about 40%, recurrences are frequently observed. Metastases develop within the first 24 months [323] and are found in the lung, liver, kidneys, and bones [321]. The overall prognosis is poor and reveals an overall survival rate of about 60% [106].
2.4.2.3.6 Biphenotypic sinonasal sarcoma (BSS)
Biphenotypic sinonasal sarcomas have been included in the WHO classification of tumors of the sinonasal system of 2017 as a new entity. This is justified by the defined PAX3/MAML3 translocation that makes it an independent entity. Up to now, the malignoma was wrongly classified as leiomyosarcoma [325]. Up to now, BSS has only been described in the sinonasal tract.
BSS is a low-grade spindle cell sarcoma. It occurs preferably in females (f:m=3:1) and affects people in an age span between 24 and 85 years with a mean age of 52 years [106].
Predominant manifestation sites are the sphenoid cells (57%) and the nasal cavity (54%), often with transgressing growth [326] and also infiltration of the orbita or olfactory fossa.
The symptoms are unspecific. Patients report about nasal obstruction, pressure sensation, and facial pains. Macroscopically, a polypoid, partly solid, reddish and grey tumor mass is seen. Microscopically, the cellular structure has focal cell-dense parts in the tumor that are similar to adult fibrosarcoma with superficial hamartoma-like proliferation of included, local mucosal glands. Focally, ectatic vessels with bleeding and hemangioperycytoma-like aspect are frequently found [326].
Due to the fact that the malignoma was described for the first time in 2012 [327] and included in the WHO classification only in 2017 as independent entity, it is mostly unknown, however, it might occur more frequently than assumed because many cases have been classified as malignant peripheral nerve sheath or muscular tumors.
Slow growth and local infiltration of neighboring structures are characteristic for BSS. About 50% of the patients of the originally described cohort have local recurrences within a period of 9 years. Distant metastases have not been reported up to now [327]. In addition, only one lethal outcome has been documented for this disease [328].
The therapy of choice consists of tumor resection with sufficient safety margins such as for other sarcomas. Adjuvant radio- and chemotherapy can be applied in cases with advanced tumor stage. The efficiency of adjuvant radio- and chemotherapy is unknown due to the very low number of cases [329] [330].
2.4.2.3.7 Malignant peripheral nerve sheath tumor/neurofibrosarcoma
Malignant peripheral nerve sheath tumors (MPNST) develop from peripheral nerves or the transformation of benign tumors of peripheral nerves. Generally, they have a differentiation to one of the components of nerve sheaths (e. g. schwann cells [malignant schwannoma], fibroblasts, or perineural cells).
The malignomas occur mainly in adult patients with a large age range and a peak within the 5th decade of life. In about 20–25% of the cases, the malignant peripheral nerve sheath tumor is associated with type 1 neurofibromatosis. In these cases, the patients are often younger (between 30 and 40 years) [106] [331]. An association with previous radiotherapy is discussed [332].
In patients with type 1 neurofibromatosis, the incidence amounts to 1:3,500. Regarding the total population, the incidence amounts to 1:100,000 [333] [334]. About 20% of all malignant peripheral nerve sheath tumors are located in the head and neck. Sinonasal manifestation is much more rarely observed.
Manifestation sites are located along the cerebral nerves, mainly the vestibular and vagus nerve [106] [331].
Clinical symptoms of affected patients are rapidly progressive painful swellings and neurological deficits of the affected nerves.
Malignant peripheral nerve sheath tumors are not encapsulated and have a highly infiltrative growth. Most different cellular morphologies are found, among them spindle-shaped, epitheloid and pleomorphic cells). Histologically, the difference is made between spindle cell and glandular malignant peripheral nerve sheath tumors that are different with regard to their fascicle-like alignment or the presence of goblet cells [106]. The malignomas are classified into low- or high-grade entities according to the mitosis rate, the presence of atypical mitoses, pleomorphism, and necrosis [332].
In general, these tumors are very aggressive and have a poor prognosis. Tumor excision with sufficient safety margins is the therapy of choice. The majority of malignant peripheral nerve sheath tumors are high-grade sarcomas with a tendency of local recurrence and distant metastasis. 40–65% of the affected patients reveal local recurrences, distant metastasis develops in 30–60% of the cases, mainly in the lung, liver, brain, bone, and adrenals. Regional lymph node metastasis is rare so that neck dissection should not be performed as standard [333]. Adjuvant radiotherapy, if needed in combination with chemotherapy, has a positive impact on the 5-year survival (65 vs. 38%) [335].
2.4.2.3.8 Sinonasal synovial sarcoma
Synovial sarcomas are mesenchymal tumors with variable epithelial differentiation and gland formation. Typically, a specific chromosomal translocation (t(X;18) (p11:q11)) is observed that leads to the development of an SS18-SSX fusion gene [106] [336].
The synovial sarcoma is the most frequent non-rhabdomyosarcoma soft part sarcoma in children, adolescents, and young adults with an age span between the 3rd and 4th decade of life.
The etiology is strongly associated with previous radiotherapy [337] [338] [339]. Manifestation in the sinonasal tract or the skull base occurs extremely rarely.
Clinically, a palpable, mostly deeply located mass is found with or without pain on pressure. Macroscopically, the tumor is yellow to grey or whitish. Slowly growing synovial sarcomas are generally well delimited. Microscopically, monophasic (spindle-cell shaped, calcifying/ossifying, myxoid, and poorly differentiated) as well as biphasic subtypes with glandular or solid epithelial cell may be differentiated. Poorly differentiated tumors may contain areas with frequent mitosis and necrosis [106].
Within the last decades, the therapy has only changed unimportantly and consists of tumor resection for circumscribed findings, often combined with radio- or chemotherapy. Combined therapies are applied depending on the stage of the disease. According to a large trial, stage I and II tumors undergo resection with postoperative radiotherapy, in stage II, tumor resection is combined with radiochemotherapy [340]. Neoadjuvant chemotherapy may be applied in patients with locally advanced tumor growth where otherwise mutilating surgery would be required. First results of immunotherapy showing an activity for NY-ESO-1, trabectedin and a multitude of angiogenesis inhibitors are promising [341].
2.4.2.4 Borderline and low-malignant entities of the soft tissue
2.4.2.4.1 Aggressive fibromatosis of the desmoid type
Aggressive fibromatosis is a clonal spindle-cell neoplasm with infiltrative growth. Distant metastases do not occur. The terms of desmoid fibromatosis and aggressive fibromatosis are used as synonyms of aggressive fibromatosis of the desmoid type.
The incidence is estimated to 1:250 000 to 1:500 000. The ages of disease onset reach from 15 to 60 years, about 30% of the cases develop in childhood. The manifestation in the head and neck concern about 15% of all aggressive fibromatoses. The sinonasal manifestation is even rarer [106] [342] [343].
The etiology is still unknown. An association with Gardner’s syndrome (see 2.4.1.2.1) and the familial colorectal polyposis is possible [344] [345].
Aggressive fibromatoses of the desmoid type show poorly delimited lesions with focally infiltrative growth that macroscopically present as solid, whitish lesions with trabecular pattern. Immunohistochemistry of desmoids shows nuclear staining with β catenin and additionally cytoplasmatic background [326]. Fascicular growth with spindle-shaped cells and mild nuclear pleomorphisms may be visualized. Atypical mitoses and necroses are not present. The stroma can be variably collagenized and myxoid or mucous.
Adequate paint therapy is required in patients with aggressive fibromatosis. Emori et al. could show in a series of 16 cases that the pain accompanying desmoid tumors is associated with an overexpression of cyclo-oxygenase 2 [346].
Generally, the complete surgical resection is the therapy of choice. Due to the anatomical proximity to critical structures, a systemic drug-related therapy (anti-estrogen therapy, non-steroidal antiphlogistics), chemotherapy (vinblastine/vinorelbine, pegylized liposomal doxorubicin), tyrosine kinase inhibitors (e. g. imatinib, sorafenib), or radiotherapy may be applied if the lesion is located in the sinonasal tract [326] [343] [347]. Within the last years, however, more frequently a wait-and-see strategy was pursued because available data show that only a small percentage of aggressive fibromatoses progress [348] which occurs within the first 36 months after diagnoses [349].
The prognosis is good in cases of R0 resection. In cases of R1 resections, recurrences usually develop within less than 2 years [350].
2.4.2.4.2 Sinonasal glomangiopericytoma
Sinonasal glomangiopericytomas are spindle cell neoplasms that have been included in the WHO classification of head and neck tumors as new entity. Beforehand, this entity was classified as sinonasal hemangiopericytoma. Within the last 60 years, the term of hemangiopericytoma was used in order to describe a multitude of neoplasms that have similar morphological properties. These characteristics concern about 15% of all soft tissue neoplasms [351] which leads to confusion for establishing a specific tumor regime [352]. Today, the term of hemangiopericytoma does not describe an independent neoplasm but rather a growth pattern that is found in several neoplasms that are very different [353].
Glomangiopericytomas represent about 0.5% of all sinonasal neoplasms. Up to now about 100 cases have been described [352]. The age peak is in the 7th decade of life with a slight predilection of female patients [106].
The most frequently observed manifestation site is the nasal cavity with extension into the adjacent paranasal sinuses. Isolated cases in the sinuses are rare. Glomangiopericytomas mostly develop only on one side.
Affected patients report about nasal obstruction with pressure sensation and epistaxis. The duration of the complaints until diagnoses often lasts for more than one year [354].
Macroscopically, the tumor shows a polypoid, red to pink appearance. The surface is soft and fleshy. On the average, the tumor measures 3 cm at the time of diagnosis. Microscopically, an unencapsulated growth is observed under an intact epithelial layer with only rare erosions especially in large tumors. A pattern-less diffuse structure with partially fascicular cell arrangement is characteristic, separated from the vascular plexus of capillaries up to large caverns and a prominent acellular hyalinization [106]. Cellular atypia is not found. In contrast to demoids (see 2.4.2.4.1), B catenin can be identified in nearly all tumor cells [326].
Glomangipericytomas grow slowly and have a very good survival rate. Recurrences occur in up to 40% of the cases, mostly as a consequence of incomplete resection. Invasive growth behavior is observed in cases of tumors larger than 5 cm [354] [355].
Despite the low number of cases, tumor resection is considered as standard therapy. Radio- and chemotherapy may be applied in the context of unresectable tumors or distant metastasis. Adjuvant radiotherapy is possible in order to improve the local tumor control. Depending on the degree of vascularization, preoperative embolization is recommended to reduce the intraoperative blood loss [356] [357] [358].
2.4.2.4.3 Sinonasal solitary fibrous tumor
Solitary fibrous tumors originate from a fusion of the genes NAB2 and STAT6 and have a fibroblastic phenotype with intensively branching vascular structures.
As synonyms, the terms of hemangiopericytoma or giant cell angiofibroma are used.
Solitary fibrous tumors are a rarity and responsible for less than 0.1% of all sinonasal neoplasms. They affect mainly adults without clear predilection for one gender [106].
The tumors mostly develops in the nasal cavity and leads to nasal obstruction and epistaxis as well as unspecific complaints such as pressure sensation.
Macroscopically, a polypoid, solidly structured whitish tumor is seen that is usually small because of the limited space in the sinonasal tract [359] [360]. Histologically, pseudo-encapsulated tumors lying under the mucosa are found with variable cell formations, among them spindle-shaped cell formations that seem to be arranged randomly. The vessels are arranged radially or antler-like [106] [325].
The therapy of choice is the complete tumor resection which usually leads to a cured condition. In patients older than 55 years, a tumor size of more than 15 cm, necrotic tumor areas, and more than 4 mitoses per 10 high-resolution fields, a more aggressive growth seems to be obvious [361] [362]. Adjuvant radiotherapy seems to be an additional option in these cases, in particular in cases of incomplete resection and local recurrences [363]. The efficiency of this therapy, however, cannot be finally assessed due to the extremely low number of cases.
2.4.2.4.4 Epitheloid hemangioendothelioma
Epitheloid hemangioendotheliomas are low- to intermediate-grade neoplasms from cells showing an endothelial phenotype, epitheloid morphology, and a hyalinized, chondroid, or basophil stroma [106].
They affect in particular adults. Due to their extremely rare occurrence and the complex differential diagnostics, an estimation of the incidence is difficult.
Their occurrence in the head and neck is very rare. Their origin is mostly in the soft tissue, the skin or bone. Extremely rarely, lymph nodes are seen as primary manifestation sites [364] [365] [366] [367].
Epitheloid hemangioendotheliomas grow slowly, infiltrate surrounding structures and rarely metastasize [368]. The symptoms are mostly unspecific. Depending on the location, epistaxis may occur.
Macroscopically, a nodular tumor mass with pale, partly reddish, partly hemorrhagic cutting surface is found. Histologically, endothelial and histocytic cells are seen that are arranged in short, fibrous formations in a myxohyalinic stroma. The mitotic activity is low. Multicellular vascular channels may be present. Endothelial markers can be identified (CD31, ERG, FLI1). In most cases is a gene fusion of WW-TR1-CAMTA1 is found [106].
Most cases have an indolent course. Care reports with tumor-related mortality are available [369] [370] [371].
The therapy of choice is the radical tumor resection [372] that leads to a recurrence-free situation in up to 85% [373]. Drug therapy may be performed as curative or adjuvant treatment. It comprises a combination of corticosteroids, cytotoxic agents, thrombocyte aggregation inhibitors, and antifibrinolytics, and interferon-alpha. In cases where R0 resection cannot be achieved because of the high morbidity, additional radiotherapy may be discussed [373] [374].
2.4.2.5 Hematolymphoid tumors
2.4.2.5.1 Extranodal NK/T cell lymphoma
The extranodal NK/T cell lymphoma is an extranodal lymphoma with cytotoxic phenotype and mandatory association with the Epstein-Barr virus. The terms of angiocentric lymphoma and lethal midline granuloma are used synonymously.
The malignoma has a higher prevalence in Southeast Asia and the indigenous population of Mexico and Central as well as South America. In these areas, it represents up to 10% of Non-Hodgkin lymphomas. In contrast, this percentage in North America and Europe amounts to less than 1%. The prevalence of the disease in Europe is estimated to less than 9:1 000 000 people [375].
The extranodal NK/T cell lymphoma grows in a destructive way in the upper aerodigestive tract and manifests in the nasal cavity, the paranasal sinuses and along Waldeyer’s tonsillar ring [106].
First symptoms are nasal obstruction and epistaxis. The infiltrative growth often leads to perforation of the nasal septum of the hard palate and to infiltration of the skin where ulcerating lesions develop at the penetration sites. If the lesion is located in the paranasal sinuses, symptoms of chronic rhinosinusitis may mask the actual disease [106]. Functional impairment is observed in cases of ocular or cerebral infiltration.
Histopathologically, a diffuse tumor infiltrating the tissue with angiocentric or angioinvasive growth pattern and large necrotic areas is seen. The neoplastic cells vary with regard to their size and number of irregularly shaped nuclei. Immunohistochemically, the tumor expresses CD3, cytotoxic markers, and CD56 [376] [377].
Due to the strict EBV association and the clear ethnical predisposition, a gene defect of the immune response of affected patients with regard to EBV infection is assumed [378] [379].
In comparison to other T cell lymphomas, the prognosis is poor with a median survival rate of 7.8 months and a 5-year survival rate of 40% [15]. The plasmatic EBV DNA load is highly significant regarding the diagnosis and prognosis. It was integrated in the prognostic algorithm together with PET/CT results [106] [380]. Current chemotherapy regimen achieve long-term remissions in 70–80% of the cases with stage I/II and about 50% with stage III/IV [381] [382].
2.4.2.5.2 Extraosseous plasmocytoma
Extraosseous plasmocytomas are proliferations of monoclonal plasma cells with extraosseous manifestation without the basic multiple myeloma. It is important to differentiate them from B cell lymphomas with plasmocytic/plasmoblastic differentiation, especially from MALT lymphomas and plasmoblastic lymphomas [106].
The median age of disease onset at the time of diagnosis is about 60 years, and there is an increased prevalence for male patients (m:w=3–4:1) [106] [383] [384].
About 80% of extraosseous plasmocytomas manifest within the upper airways, mainly the nasal cavity and the paranasal sinuses. The worldwide incidence is estimated to 0.021 to 4 per 100 000 people [385].
Clinically, solitary masses are found that lead to nasal obstruction, epistaxis, facial pain, chronic sinonasal complaints with rhinorrhea and depending on the infiltration to cerebral deficits and bulbar protrusion.
Less than 25% of the patients have a monoclonal serum paraprotein, typically of the IgA type. Diagnostic characteristics of multiple myeloma are not found [386] [387].
Histologically, a diffuse infiltration of well, moderately, or only poorly differentiated plasma cells and sometimes amyloid deposits are found. The difference must be made between moderately and well differentiated extraosseous plasmocytomas and B cell lymphomas, in particular of MALT lymphomas with extensive plasmocytic differentiation. Poorly differentiated extraosseous plasmocytomas must be delimited of plasmoblastic lymphomas [388] [389]. The cells often express characteristics of plasmocytic differentiation (C138, C38, VS38, MUM1/IRF4) [390]. Monotypic immunoglobulin light chains can typically be identified.
The therapy of choice is local radiotherapy which leads to a significantly better prognosis than in cases of multiple myeloma. The disease-specific 5- and 10-year survival rates amount to 82 and 76%, respectively, according to the current literature [391] [392] [393]. Local recurrences or dissemination into other extraosseous locations are possible. About 15% of the patients develop multiple myeloma in the course of the disease [383].
2.4.2.5.3 Langerhans cell histiocytosis
In the context of Langerhans cell histiocytosis (LCH), proliferation and accumulation of Langerhans cells is observed in various tissue types. The prevalence in the European population is estimated to 1–2:100 000. Primary manifestation of LCH in the nose, the sinuses, or at the skull base is seen even more rarely. Exact data do not exist. The disease develops frequently in children. The age peak of affected patients is between 1 and 4 years [394] [395]. Typical manifestations of LCH are found in the bone (80%), the skin (35%) as well as the pituitary gland (25%). More rarely, organ systems such as lung, liver, and hematopoetic system (15–20%) are affected [396]. Severe courses may lead to sclerotic cholangitis or neurodegenerative changes in up to 2% of the cases.
If the nose or the paranasal sinuses are affected, mostly pressure sensation and pains of the affected regions are identified. In cases of affected frontal or maxillary sinus, swellings in the face may occur [397]. Due to the space-requiring effect of the manifestation of LCH, affections of neighboring structures such as the optic nerve are possible [398].
Imaging of the disease shows well delimited bone lesions that nearly seem “punched out” and involve the adjacent soft tissue. Lesions without bony erosions could also be found [399] [400]. In an analysis of 163 patients with LCH, MRI showed an involvement of the paranasal sinuses or the mastoids in 55% of the cases [401].
Recent trials showed that patients with disseminated disease had a BRAF V600E mutation that is also found in malignant melanomas [402] [403] [404]. The option of applying a targeted therapy with inhibitors that are already used in the context of malignant melanoma therapy, is currently investigated. Not least because of this mutation, the discussion persists if LCH is a malignant disease with varying clinical manifestation.
Due to the not finally analyzed pathogenesis of LCH, the therapy is empiric and depends on the respective manifestation and the degree of systemic affection. The diagnosis is confirmed by means of biopsy. The excision or curettage of the affected areas can be an effective treatment of unifocal manifestations [405]. In cases of unifocal manifestation, the prognosis is good. The application of 125 mg methylprednisolone is recommended due to the inhibitor effect on the osteolysis. Radiotherapy is mainly applied in cases of recurrences. The prognosis is significantly poorer when systemic affection with multifocal extent is diagnosed. For therapy, drug therapy with vinblastine, prednisone, etoposide, and methotrexate are applied in various combinations [406].
2.4.2.6 Neuroectodermal and melanocytic tumors
2.4.2.6.1 Ewing sarcoma, primitive neuroectodermal tumor
Ewing sarcomas and primitive neuroectodermal tumors are primitive, round/small cell high-grade sarcomas with variable neuroectodermal differentiation. Translocation between the WESR1 gene and chromosome 22 as well as a member of the ETS transcription family is characteristic [106].
The terms of peripheral neuroectodermal tumor, peripheral neuroepithelioma, peripheral neuroblastoma, or adult neuroblastoma are used synonymously.
Ewing sarcomas and primitive neuroectodermal tumors manifest in only 2–10% of the cases in the head and neck and have a higher incidence in males. Predominantly children and young adults are affected [106].
Manifestation sites in the head and neck area are the skull and jaw bones, clearly less frequently the paranasal sinuses or the nasal cavity are affected. Most frequent location of the sinonasal tract is the maxillary sinus and the nasal cavity. Infiltrations into the orbita or in intracranial direction are possible [407] [408]. [Figure 15] shows MR imaging of the skull of a 9-year old patient who presented with left-sided paresis of the abducens nerve.

Pains, progredient swellings, rapidly progressive nasal obstruction are typical symptoms of affected patients.
Macroscopically, polypoid or multilobular, grey/whitish, partly hemorrhagic tumors with ulcerations are observed. Histologically, a clearly cellular tumor with diffuse to lobular growth and trabecular or strain-like structure is seen. Uniform small cells with round to oval nuclei, discrete nuclear chromatin, pale cytoplasm and unclear cell borders are additional characteristics. The mitotic activity is variable with 5–10 mitoses per high-resolution field [106]. In 90–95% of the cases, a consistent, reciprocal translocation between the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 is identified [409].
Beside RMI and CT scan, PET/CT is recommended for staging in order to exclude distant metastasis or to correctly locate it so that possible surgical procedures may be planned adequately [410]. If possible, excision of the findings with sufficient safety margins should be intended. Even if Ewing sarcomas are supposed to be sensitive on radiation, the number of patients who underwent primary radiotherapy within the last 30 years has continuously decreased. One reason might be the late toxicity and the risk of secondary malignomas after radiotherapy because mainly children are affected by the disease. The modification of chemotherapies within the last decades significantly increased the survival rates of originally 10%. The current 5-year survival rate for sinonasal Ewing sarcomas/primitive neuroectodermal tumors without metastatic spread amounts to 50–75% and is better than for other locations [408] [411]. Chemotherapy schemes include VACA (vincristine, doxorubicin, cyclophosphamide, actinomycin), VAIA (ifosfamid instead of cyclophosphamide) or EVAIA (additional etoposide), and VIDE (without actinomycin). Despite the promising development of the prognoses, the outcome for patient with metastases is clearly poorer [412].
2.4.2.6.2 Olfactory neuroblastoma
Olfactory neuroblastomas are malignant neuroectodermal neoplasms. According to their genesis, they are frequently located in the cranial parts of the nasal cavity along the olfactory region. The term of esthesioneuroblastoma is used synonymously.
Their incidence amounts to about 4:10 000 000. Overall, this entity represent about 3% of all malignomas of the paranasal sinuses [106] [413]. The age of the affected patients reaches from 2 to 90 years in the cases described in the literature with an age peak between the 5th and 6th decade of life. Males are more frequently affected (1.2:1). Ethnical or familial predilection does not exist.
The tumor may develop along the lamina cribrosa, the medial part of the middle turbinate, or the cranial part of the nasal septum. Originating locations are the vomeronasal organ, the sphenopalatine ganglion, according to the embryology the olfactory placode, or the terminal nerve that fills the ethmoid cleft of the anterior part of the lamina cribrosa [414]. Ectopic manifestations in the sphenoid sinus are possible, however, they are extremely rare for all other sinuses [415] [416]. Due to the point of origin, the skull base is generally involved in the tumor development.
Initially, olfactory neuroblastomas often cause complaints that are similar to benign lesions so that diagnosis is often made rather late. Nasal obstruction and occasional epistaxis are reported. Cephalgia, rhinorrhea, epiphora, and visual disorders are symptoms of already advanced tumor growth. Anosmia is rare with less than 5% despite the location at the lamina cribrosa. Paraneoplastic syndromes occur in about 2% of the cases.
Clinically, a tumor mass is observed that originates from the lamina cribrosa and extends over large parts. Mostly the tumor grows unilaterally and appears polypoid, soft with red-grey surface and intact mucosa. Computed tomography and especially magnet resonance imaging allow determining the extension ([Figure 16]) and depiction of an infiltration of the orbita which can be seen in [Figure 17].


Several staging systems are available (Kadish [417], Morita [418], and according to TNM [419] while the classification according to Kadish is mostly used.
Olfactory neuroblastomas are classified into low-grade and high-grade entities. Low-grade olfactory neuroblastomas show sharply demarcated, submucously growing cellular nests that are often separated by vascular or hyalinized connective tissue. So-called Homer Wright rosettes/pseudo-rosettes where neoplastic cells surround in a palisade-like way the central neural matrix are characteristic. High-grade olfactory neuroblastomas show necrotic tumor areas, pleomorphisms, an increased mitotic rate as well as a less apparent lobular growth [106].
The mostly known grading system was established by Hyams et al. [420]. Hereby the difference is made between grade I (highly differentiated) to grade IV (poorly differentiated) lesions. The criteria are the tumor architecture, mitotic activity, nuclear polymorphisms, fibrillary matrixes and rosettes, necrotic areas, glandular proliferation, and calcifications.
Mao et al. could show in an article published in 2009 that the PTCH1, GLI1, and GLI2 signaling pathways are involved in the pathogenesis of olfactory neuroblastomas so that a central role of the sonic hedgehog signaling pathway can be assumed [421].
Primary therapeutic option is the tumor resection, followed by local radiotherapy. In contrast to the traditional craniofacial resection that was first described in 1963 [422], today the endoscopic tumor resection is favored. Originally it was assumed that only en-bloc resection of the tumor may secure long-term recurrence-free conditions, however, endoscopic procedures show similarly favorable outcomes with clearly less invasive interventions [423]. Advanced tumor growth may require additional chemotherapy. High-grade tumors according to Hyams show better responses so that in individual cases also induction chemotherapy can be discussed [424].
Dulguerov et al. analyzed in a meta-analysis the courses of the disease in dependence of the therapeutic schemes and revealed the best outcome for patients who had undergone tumor resection followed by local radiotherapy. Patients of this group had a 2-year survival rate of 65% compared to 48% and 37%, respectively, for tumor resection alone and radiotherapy alone [425]. Current study results further show a significantly improved disease-specific survival for patients with stage T3/T4 who received tumor resection and radiotherapy compared to surgical therapy alone [426].
2.4.2.6.3 Malignant mucosal melanomas
Mucosal melanomas are malignant neoplasms that develop from melanocytes of the mucosa and are biologically different from cutaneous melanomas. Their etiology is unknown.
Mucosal melanomas are responsible for less than 1% of all malignant melanomas [427] and represent about 4% of all sinonasal tumors. The age span is rather broad and has a peak within the 7th decade of life [106].
Most frequent manifestation site is the nasal cavity with the nasal septum and clearly more rarely the nasopharynx or the maxillary sinus [428] [429]. Due to the missing exposure to sun light, the sinonasal mucosal melanoma seems to have a different biology compared to the cutaneous malignant melanoma. About 20% of the affected patients have multifocal foci at the time of diagnosis, about 40% of the diagnosed melanomas are amelanotic [430].
Malignant mucosal melanomas may present as plane, pigmented mucosal changes ([Figure 18]) or as polypous massive tumor formation ([Figs 19] and [20]). Symptoms at first presentation are often epistaxis and nasal obstruction. Such unspecific complaints often lead to a diagnosis in higher tumor stages.



Macroscopically, mucosal melanomas have a polypoid structure. They may be pigmented and fragile, light brown or grey or solidly configured [106]. Microscopically, often ulcerations are found with mucosal covering with variable cellular morphology that reaches from epitheloid/undifferentiated cells to spindle-like, plasmacytoid and rhabdoid cells with partly prominent nuclei. Atypical mitoses are frequently observed. Nearly half of the malignomas can show amelanotic manifestations which clearly increases the number of possible differential diagnoses (e. g. olfactory neuroblastoma, rhabdomyosarcoma, sinonasal undifferentiated carcinoma, poorly differentiated squamous cell carcinoma) [106].
In contrast to cutaneous melanomas as well as choroidal melanomas, higher rates of KIT and NRAS mutations are found. BRAF mutations, however, are rare [431] [432].
It is decisive for the staging and the prognosis to correctly differentiate mucosal melanoma from sinonasal metastasis of a cutaneous malignant melanoma. Distant metastasis and advanced patient age are the most decisive prognostic factors. According to the 7th issue of the “Cancer Staging Manual of the American Joint Committee on Cancer”, a very unfavorable 5-year survival rate of less than 30% is seen in cases of stage T3 and T4 tumors [433] [434] [435].
Current treatment recommendations favor the wide local excision if possible [436]. The role of postoperative radiotherapy is controversially discussed. A meta-analysis from 2012 could not reveal higher survival rates [437]. Other study results confirm an increased 3-year survival rate from 18 to 30% as well as a reduced local recurrence rate after postoperative radiotherapy [438] [439]. Treatment with anti-PD-1 antibodies as well as anti-CTLA-4 antibodies show promising results in advanced or metastasized mucosal melanomas, however, the evidence is too low to justify their clinical application [436] [440].
2.4.2.7 Germ cell tumors
2.4.2.7.1 Sinonasal terato-carcino-sarcoma
Terato-carcino-sarcomas are malignant neoplasms with histological characteristics of teratomas as well as carcino-sarcomas without malignant germ cell parts. The terms of malignant teratoma, blastoma, terato-carcinoma, and teratoid carcino-sarcoma are used as synonyms.
Terato-carcino-sarcomas are very rare tumors that mainly occur in male adults (mean patient age of 60 years).
Most frequently, the tumor develops in the nasal cavity followed by the ethmoid and maxillary sinus. In about 20% of the cases, intracranial involvement can be found [441].
The diagnosis is made based on the evidence of malignant epithelial elements and two or more malignant mesenchymal components such as for example fibroblasts, cartilage, bone, or smooth muscles. These combinations may be similar to primitive bronchial or intestinal structures that seem to be strange in the sinonasal tract [442]. These teratoid elements are significant prognostic factors of the sinonasal terato-carcino-sarcoma. Fetal, clear-cell squamous epithelium and tubular or glandular structures are further criteria for the diagnosis. Stem cell parts are not found in terato-carcino-sarcomas [443–447].
The treatment of the sinonasal terato-carcino-sarcoma is difficult due to the high malignancy rate and the poor prognosis. In order to have realistic chances for long-term tumor-free survival, radical tumor resection is required followed by radiotherapy [448]. In cases of intracranial extension, combined intra- and extracranial approaches have been described to achieve en-bloc resection [449]. A review article published by Misra et al. identified 5 reports supporting endoscopic tumor resection [441]. The role of adjuvant chemotherapy is not confirmed due to the low number of case reports. Because of the extremely rare occurrence, the characteristics and optimal therapeutic schemes are not defined.
Lymph node and distant metastasis are frequently observed. The reported survival rates vary from 50–70% in different analyses [106, 441].
Summary
The review article presents rare diseases of the nose, the paranasal sinuses as well as the anterior skull base with focus on malformation, ventilation and functional disorders and tumor diseases. Due to the complexity of the anatomic region and the multitude of possible diseases (functional disorders, malformations, tumors), this presentation does not claim to be complete.
In many cases, the adequate therapy of a rare disease is complex and requires extensive research of experiences described in the literature. Especially in cases of benign and malignant neoplasms, the use of adjuvant therapy is often not clarified and needs experience that is not available in many centers because of the low number of cases. Also within the group of rare diseases, a subdivision has to be performed because extreme differences exist regarding the incidences of the single diseases. An incidence of less than 5:10 000 fulfills the criteria of the definition of an orphan disease, but in a large center of oto-rhino-laryngology, head and neck surgery, it may lead to a remarkable number of cases. Diseases for which less than 100 case reports are available are a challenge also for large centers.
Thus it seems to be reasonable to establish competence centers for certain rare diseases. Furthermore, intensive exchange between the centers is essential with regard to the collected data in order to develop diagnostic and therapeutic possibilities for rare diseases. The structural law of hospitals from 2015 which regulates among others financial allowances for centers for orphan diseases, also defines their tasks comprising the establishment of registries or atlases of care supply so that affected patients may retrieve information. Especially for rare diseases, registry trials are a significant basis for data collection and future statistical analysis regarding therapeutic benefit, quality of life, costs of therapy as well as drug safety. In the near future, also patients suffering from rare diseases of the nose, the paranasal sinuses, and the anterior skull base may benefit. ([Tables 3] and [4]).
|
Entity |
Number of cases |
Percentage |
|---|---|---|
|
Pleomorphic adenoma |
73 |
23% |
|
Oncocytic tumor |
7 |
2% |
|
Low-grade adenocarcinoma (including acinar cell carcinoma) |
67 |
21% |
|
Mucoepidermoid carcinoma |
17 |
5% |
|
Adenoid cystic carcinoma |
54 |
17% |
|
High-grade adenocarcinoma |
93 |
30% |
|
Total |
311 |
100% |
|
Staging according to Kadish |
|
|---|---|
|
A |
Tumor limited to the nasal cavity |
|
B |
Tumor extends to the paranasal sinuses |
|
C |
Tumor extends to the cribriform plate, skull base, orbita, or intracranial cavity |
|
D (addition according to Morita [417]) |
Cervical or distant metastases |
Interessenkonflikt
Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
Acknowledgement
The author expresses his thankfulness to Prof. Dr. Thomas Hoffman and Prof. Dr. Jörg Lindemann for critically reviewing the manuscript as well as Prof. Dr. Wolfgang Pirsig for the figures provided and the impulses given regarding the chapter on malformations of the nose. Further, the author thanks Prof. Dr. Meinrad Beer, radiologist at the University Hospital of Ulm for the CT scans and MR imaging provided for perusal. He also thanks Ms. Alexandra Gey, Prof. Dr. Stefan Plontke, and Dr. Alexander Glien (Department of Oto-Rhino- Laryngology, University Hospital of Halle) for providing the Figures 13 and 14 as well as Prof. Dr. Sabrina Kösling (Department of Radiology, University Hospital of Halle).
-
Literaturverzeichnis
- 1 Bundesministerium für Gesundheit Seltene Erkrankungen - Bundesgesundheitsministerium.
2019 Im Internet: https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
MissingFormLabel
- 2 Anzahl I. Prävalenzen en und Inzidenzen seltener Krankheiten: Bibliografische Angaben. 2015; 1-52 . Im Internet: https://www.orpha.net/orphacom/cahiers/docs/DE/Pravalenzen_seltener_Krankheiten_Alphabetische_Liste.pdf
- 3 Pedan H, Janosova V, Hajtman A, Calkovsky V. Non-Reflex Defense Mechanisms of Upper Airway Mucosa: Possible Clinical Application. Physiol Res 2020; 69 (Suppl. 01) S55-S67 PMID: 32228012
- 4 Sommer F, Scheithauer M, Kröger R. et al. Niesen als mechanische Abwehr – Numerische Simulation mit Analyse der durchströmten Nasenbereiche. Laryngorhinootologie 2014; 93: 746-750
- 5 Sommer F, Hoffmann TK, Mlynski G. et al. Dreidimensionale Analyse nasaler PhysiologieThree-dimensional analysis of nasal physiology. HNO 2017; 66: 280-289
- 6 Lindemann J, Reichert M, Kröger R. et al. Numerical simulation of humidification and heating during inspiration in nose models with three different located septal perforations. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1795-1800
- 7 Sommer F, Kröger R, Lindemann J. Numerical simulation of humidification and heating during inspiration within an adult nose. Rhinology 2012; 50: 157-164
- 8 Lindemann J, Rettinger G, Kröger R. et al. Numerical simulation of airflow patterns in nose models with differently localized septal perforations. Laryngoscope 2013; 123: 2085-2089
- 9 Götte K, Hörmann K. Sinonasal malignancy: What’s new?. Orl 2004; 66: 85-97
- 10 Barnes L, Eveson JW, Reichart P. et al. World health organization classification of tumours: Pathology and genetics of head and neck tumours. Lyon: International Agency for Research on Cancer; 2005
- 11 Slinger CA, McGarry GW. Nose and sinus tumours: Red flags and referral. Br J Gen Pract 2018; 68: 247-248
- 12 Le QT, Fu KK, Kaplan M. et al. Treatment of maxillary sinus carcinoma: A comparison of the 1997 and 1977 American Joint Committee on Cancer staging systems. Cancer 1999; 86: 1700-1711
- 13 Tiwari R, Hardillo JA, Mehta D. et al. Squamous cell carcinoma of maxillary sinus. Head Neck 2000; 22: 164-169
- 14 Tiwari R, Hardillo JA, Tobi H. et al. Carcinoma of the ethmoid: Results of treatment with conventional surgery and post-operative radiotherapy. Eur J Surg Oncol 1999; 25: 401-405
- 15 Laudien M. Ausgewählte seltene rhinologische Krankheitsbilder Pathogenese – Klinik – Therapie. Laryngorhinootologie 2015; 94: S272-S287
- 16 Hauser LJ, Jensen EL, Mirsky DM. et al. Pediatric anosmia: A case series. Int J Pediatr Otorhinolaryngol 2018; 110: 135-139
- 17 Hummel T, Whitcroft KL, Andrews P. et al. Position paper on olfactory dysfunction. Rhinol J 2017; 0: 1-30
- 18 Orphanet: Anosmie, isolierte kongenitale. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=11807&Disease_Disease_Search_diseaseGroup=anosmie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Anosmie--isolierte-kongenitale&title=Anosmie
isolierte kongenit
MissingFormLabel
- 19 Mahmood AN, Abulaban O, Janjua A. Bilateral olfactory aplasia: Uncommon cause of congenital anosmia. Clin Case Reports 2019; 7: 1456-1457
- 20 Orphanet: Kallmann Syndrom. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=478
MissingFormLabel
- 21 Johnson VP, McMillin JM, Aceto T. et al. A newly recognized neuroectodermal syndrome of familial alopecia, anosmia, deafness, and hypogonadism. Am J Med Genet 1983; 15: 497-506
- 22 Gebril O, Abdelraouf E, Shafie M. et al. Johnson-McMillin microtia syndrome: New additional family. J Fam Med Prim Care 2014; 3: 275
- 23 Orphanet: Arrhinie, isolierte. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert=1134
MissingFormLabel
- 24 Thiele H, Musil A, Nagel F. et al. Familial arhinia, choanal atresia, and microphthalmia. Am J Med Genet 1996; 63: 310-313
- 25 Abukhalaf SA, Zalloum JS, Al Hammouri A. et al. Congenital arrhinia: A case report and literature review. Int J Pediatr Otorhinolaryngol 2020; 135: 110083
- 26 Jung JW, Ha DH, Kim BY. et al. Nasal Reconstruction Using a Customized Three-Dimensional–Printed Stent for Congenital Arhinia: Three-Year Follow-up. Laryngoscope 2019; 129: 582-585
- 27 Núñez-Villaveiran T, Frohner BB, Urcelay PR. et al. Bifid nose – A mild degree of frontonasal dysplasia. A case report. Int J Pediatr Otorhinolaryngol 2013; 77: 1374-1377
- 28 Orphanet: Bifid nose. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2457&Disease_Disease_Search_diseaseGroup=bifid-nose&Disease_Disease_Search_diseaseType=Pat&Disease(s)/groupofdiseases=Bifid-nose&title=Bifid
nose&search=Disease_Search_Simple
MissingFormLabel
- 29 Sedano HO, Michael Cohen M, Jirasek J. et al. Frontonasal dysplasia. J Pediatr 1970; 76: 906-913
- 30 Genç E, Derbent M, Ergin NT. A mild case of frontonasal dysplasia: The rhinologic perspective. Int J Pediatr Otorhinolaryngol 2002; 65: 75-83
- 31 Cohen MM. Perspectives on craniosynostosis: Sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg 2009; 20: 646-651
- 32 Orphanet: Kraniorhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=de&Expert=157832
MissingFormLabel
- 33 Rüegg EM, Bartoli A, Rilliet B. et al. Management of median and paramedian craniofacial clefts. J Plast Reconstr Aesthetic Surg 2019; 72: 676-684
- 34 Orphanet: Paramedian nasal cleft. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=17067&Disease_Disease_Search_diseaseGroup=paramediannasal-cleft&Disease_Disease_Search_diseasType=Pat&Disease(s)/groupofdiseases=Paramedian-nasal-cleft&title=Paramedian%20nasal%20cleft
MissingFormLabel
- 35 Williams A, Pizzuto M, Brodsky L. et al. Supernumerary nostril: A rare congenital deformity. Int J Pediatr Otorhinolaryngol 1998; 44: 161-167
- 36 Orphanet: Polyrhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17033&Disease_Disease_Search_diseaseGroup=Polyrhinie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Polyrhinie&title=Polyrhinie&search=Disease_Search_Simple
MissingFormLabel
- 37 Orphanet: Nasenlöcher, überzählige. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17034&Disease_Disease_Search_diseaseGroup=Nasenloch-&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Nasenlocher--uberzahlige&title=Nasenl%F6cher,%FCberz%E4hlige
MissingFormLabel
- 38 Gulasi S, Turhan AH, Celik Y. et al. Accessory nostril: A rare congenital nasal anomaly. J Craniofac Surg 2015; 26: e602-e603
- 39 Galiè M, Clauser LC, Tieghi R. et al. The arrhinias: Proboscis lateralis literature review and surgical update. J Cranio-Maxillofacial Surg 2019; 47: 1410-1413
- 40 Harada T, Muraoka M. Proboscis lateralis: A rare bilateral case [7]. Ann Plast Surg 2001; 47: 350-351
- 41 Wolfe SA, Tessier P, Ciminello FS. The Arrhinias. Scand J Plast Reconstr Surg Hand Surg 2009; 43: 177-196
- 42 Rosen Z, Gitlin G. Bilateral Nasal Proboscis: Associated with Unilateral Anophthalmia, Unilateral Diffuse Pigmentation of the Conjunctiva, and Anomalies of the Skull and Brain. AMA Arch Otolaryngol 1959; 70: 545-550
- 43 El-fattah AMA, Naguib A, El-Sisi H. et al. Midline nasofrontal dermoids in children: A review of 29 cases managed at Mansoura University Hospitals. Int J Pediatr Otorhinolaryngol 2016; 83: 88-92
- 44 Orphanet: Nasal dorsum fistula. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=141219
MissingFormLabel
- 45 Jurkov M, Olze H, Klauschen F. et al. IgG4-related orbitopathy as an important differential diagnosis of advanced silent sinus syndrome. HNO 2020; 68: 65-68
- 46 MONTGOMERY WW. Mucocele of the Maxillary Sinus Causing Enophthalmos. Eye Ear Nose Throat Mon 1964; 43: 41-44
- 47 Soparkar CNS, Patrinely JR, Cuaycong MJ. et al. The Silent Sinus Syndrome: A Cause of Spontaneous Enophthalmos. Ophthalmology 1994; 101: 772-778
- 48 Naik RM, Khemani S, Saleh HA. Frontal silent sinus syndrome. Otolaryngol - Head Neck Surg (United States) 2013; 148: 354-355
- 49 McArdle B, Perry C. Ethmoid silent sinus syndrome causing inward displacement of the orbit: Case report. J Laryngol Otol 2010; 124: 206-208
- 50 Wise SK, Wojno TH, DelGaudio JM. Silent sinus syndrome: Lack of orbital findings in early presentation. Am J Rhinol 2007; 21: 489-494
- 51 Brandt MG, Wright ED. The silent sinus syndrome is a form of chronic maxillary atelectasis: A systematic review of all reported cases. Am J Rhinol 2008; 22: 68-73
- 52 Rose GE, Lund VJ. Clinical features and treatment of late enophthalmos after orbital decompression: A condition suggesting cause for idiopathic „imploding antrum“ (silent sinus) syndrome. Ophthalmology 2003; 110: 819-826
- 53 Numa WA, Desai U, Gold DR. et al. Silent sinus syndrome: A case presentation and comprehensive review of all 84 reported cases. Ann Otol Rhinol Laryngol 2005; 114: 688-694
- 54 Davidson JK, Soparkar CNS, Williams JB. et al. Negative sinus pressure and normal predisease imaging in silent sinus syndrome. Arch Ophthalmol 1999; 117: 1653-1654
- 55 Annino DJ, Goguen LA. Silent sinus syndrome. Curr Opin Otolaryngol Head Neck Surg 2008; 16: 22-25
- 56 Hildenbrand T, Klein SB, Schramek N. et al. Rare diseases of the maxillary sinus. HNO 2020; 68: 581-589
- 57 Facon F, Eloy P, Brasseur P. et al. The silent sinus syndrome. Eur Arch Oto-Rhino-Laryngology 2006; 263: 567-571
- 58 Hourany R, Aygun N, Della Santina CC. et al. Silent sinus syndrome: An acquired condition. Am J Neuroradiol 2005; 26: 2390-2392
- 59 Lee DS, Murr AH, Kersten RC. et al. Silent sinus syndrome without opacification of ipsilateral maxillary sinus. Laryngoscope 2018; 128: 2004-2007
- 60 Illner A, Davidson HC, Harnsberger HR. et al. The silent sinus syndrome: Clinical and radiografic findings. Am J Roentgenol 2002; 178: 503-506
- 61 Rose GE, Sandy C, Hallberg L. et al. Clinical and radiologic characteristics of the imploding antrum, or „silent sinus,“ syndrome. Ophthalmology 2003; 110: 811-818
- 62 Weber RK. Aktueller Stand der endonasalen Nasennebenhöhlenchirurgie
[Comprehensive review on endonasal endoscopic sinus
surgery]. Laryngorhinootologie. 2015; 94 Suppl 1; S64–S142.
doi:110.1055/s-0035-1545353. Epub 2015 Apr 10. PMID: 25860497
MissingFormLabel
- 63 Sogg A. Endoscopic sinus surgery. 4th Editio Thieme Verlag; 1993
- 64 Sommer F, Hoffmann T, Lindemann J. et al. Radicality of maxillary sinus surgery and size of the maxillary sinus ostium. HNO 2020; 68: 573-580
- 65 Thomas RD, Graham SM, Carter KD. et al. Management of the orbital floor in silent sinus syndrome. Am J Rhinol 2003; 17: 97-100
- 66 Babar-Craig H, Kayhanian H, de Silva DJ. et al. Spontaneous silent sinus syndrome (Imploding antrum syndrome): Case series of 16 patients. Rhinology 2011; 49: 315-317
- 67 Sivasubramaniam R, Sacks R, Thornton M. Silent sinus syndrome: Dynamic changes in the position of the orbital floor after restoration of normal sinus pressure. J Laryngol Otol 2011; 125: 1239-1243
- 68 Adams WM, Jones RI, Chavda SI. et al. Pneumosinus dilatans: a discussion of four cases and the possible aetiology. Rhinology 1998; 36: 40-402
- 69 Urken ML, Som PM, Lawson W. et al. Abnormally large frontal sinus. II. Nomenclature, pathology, and symptoms. Laryngoscope 1987; 97: 606-611
- 70 Kim WJ, Kim MM. Binasal hemianopia caused by pneumosinus dilatans of the sphenoid sinuses. Indian J Ophthalmol 2019; 67: 1772-1775
- 71 Skolnick CA, Mafee MF, Goodwin JA. Pneumosinus Dilatans of the Sphenoid Sinus Presenting With Visual Loss. J Neuro-ophthalmology 2001; 20: 259-263
- 72 Alatar AA, AlSuliman YA, Alrajhi MS. et al. Maxillary Pneumosinus Dilatans Presenting With Proptosis: A Case Report and Review of the Literature. Clin Med Insights Ear, Nose Throat 2019; 12: 117955061882514
- 73 Sweatman J, Beltechi R. Pneumosinus Dilatans: An exploration into the association between Arachnoid Cyst, Meningioma and the pathogenesis of Pneumosinus Dilatans. Clin Neurol Neurosurg 2019; 185
- 74 Ricci JA. Pneumosinus Dilatans: Over 100 Years Without an Etiology. J Oral Maxillofac Surg 2017; 75: 1519-1526
- 75 Martin AJ, Jarosz JM, Thomas NWM. The strange association of pneumosinus dilatans and arachnoid cyst: Case report and review of the literature. Acta Neurochir (Wien) 2001; 143: 197-201
- 76 Gibbons BA, Miele WR, Florman JE, Heilman CB, Horgan MA
Pneumosinus dilitans and meningioma: a case series and review of
the literature. Neurosurg Focus 2011; 50(5): E13. doi:10.3171/2011.
3.FOCUS1113. PMID: 21529169
MissingFormLabel
- 77 Desai NS, Saboo SS, Khandelwal A. et al. Pneumosinus dilatans: Is it more than an aesthetic concern?. J Craniofac Surg 2014; 25: 418-421
- 78 Draf W, Constantinidis J, Weber R. et al. Pneumosinus Dilatans Frontalis, Atiologie, Symptomatik Und Operationstechnik. Laryngorhinootologie 1996; 75: 660-664
- 79 Cho HS, Hong SJ, Chae HK. et al. Maxillary Sinus Pneumocele Presenting as Aesthetic Deformity: A Case Report With Literature Review. Ear, Nose Throat J 2020; 99: 397-401
- 80 Song M, Ahn SM, Reh DR. et al. Ethmoid pneumocele presenting with exophthalmos 15 years after endoscopic sinus surgery. Allergy Rhinol 2015; 6: 129-132
- 81 Braverman I. Pneumocele of the maxillary sinus with orbital and
trigeminal nerve involvement: case report and review of the
literature. J Otolaryngol Head Neck Surg 2009; 38(2): E35–E38.
PMID: 19442351
MissingFormLabel
- 82 Sasaki T, Yamoto T, Fujita K. et al. A Case of Orbital Emphysema Associated with Frontal Sinus Pneumocele. J Neurol Surg Reports 2013; 74: 054-056
- 83 Bachor E, Weber R, Kahle G. et al. Temporary unilateral amaurosis with pneumosinus dilatans of the sphenoid sinus. Skull Base Surg 1994; 4: 169-175
- 84 Bell AF, Ivan DJ, Munson RA. Barosinus pneumocele: Transient visual loss due to sphenoid sinus pneumocele in a U.S. Air Force pilot. Aviat Sp Environ Med 1995; 66: 276-279
- 85 Choi SJ, Seo ST, Rha KS. et al. Sinonasal organized Hematoma: Clinical features of seventeen cases and a systematic review. Laryngoscope 2015; 125: 2027-2033
- 86 Kim JS, Oh JS, Kwon SH. The increasing incidence of paranasal organizing hematoma: a 20-year experience of 23 cases at a single center. Rhinol J 2016; 54: 176-182
- 87 Pang W, Hu L, Wang H. et al. Organized Hematoma: An Analysis of 84 Cases with Emphasis on Difficult Prediction and Favorable Management. Otolaryngol – Head Neck Surg (United States) 2016; 154: 626-633
- 88 Song HM, Jang YJ, Chung YS. et al. Organizing hematoma of the maxillary sinus. Otolaryngol - Head Neck Surg 2007; 136: 616-620
- 89 Young D. Surgical treatment of male infertility. J Reprod Fertil 1970; 23: 541-542
- 90 Hendry WF, Knight RK, Whitfield HN. et al. Obstructive Azoospermia: Respiratory Function Tests, Electron Microscopy and the Results of Surgery. Br J Urol 1978; 50: 598-604
- 91 Arya AK, Beer HL, Benton J. et al. Does Young’s syndrome exist?. J Laryngol Otol 2009; 123: 477-481
- 92 Hendry WF, Levison DA, Parkinson MC. et al. Testicular obstruction: Clinicopathological studies. Ann R Coll Surg Engl 1990; 72: 396-407
- 93 Shiraishi K, Ono N, Eguchi S. et al. Young’s syndrome associated with situs inversus totalis. Arch Androl 2004; 50: 169-172
- 94 Hasegawa A, Fujita M, Ohe M. et al. A Rare Case of Young’s Syndrome in Japan. Intern Med 1994; 33: 649-653
- 95 Armengot M, Juan G, Carda C. et al. Young’s syndrome: A further cause of chronic rhinosinusitis. Rhinology 1996; 34: 35-37
- 96 Greenstone MA, Rutman A, Hendry WF. et al. Ciliary function in Young’s syndrome. Thorax 1988; 43: 153-154
- 97 Stern BM, Sharma G. Ciliary Dysfunction (Kartagener Syndrome, Primary Ciliary Dyskinesia). StatPearls Publishing; 2019
- 98 Samia H, Khadija B, Agnes H. et al. Long-term outcome of tunisian children with primary ciliary dyskinesia confirmed by transmission electron microscopy. Afr Health Sci 2016; 16: 954-961
- 99 Schofield LM, Duff A, Brennan C. Airway Clearance Techniques for Primary Ciliary Dyskinesia; is the Cystic Fibrosis literature portable?. Paediatr Respir Rev 2018; 25: 73-77
- 100 Alanin MC, Aanaes K, Høiby N. et al. Sinus surgery can improve quality of life, lung infections, and lung function in patients with primary ciliary dyskinesia. Int Forum Allergy Rhinol 2017; 7: 240-247
- 101 Nyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C,
Latzin P, Koerner-Rettberg C. Alternative inert gas washout outcomes
in patients with primary ciliary dyskinesia. Eur Respir J 2017; 49(1):
1600466. doi:10.1183/13993003.00466-2016. PMID: 28122863
MissingFormLabel
- 102 Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S,
Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M,
Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JE, Pohunek P,
Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A,
Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni
CE. European Respiratory Society guidelines for the diagnosis of
primary ciliary dyskinesia. Eur Respir J 2017; 49(1); 1601090.
doi:10.1183/13993003.00466-2016. PMID: PMC6054534
MissingFormLabel
- 103 Strippoli MPF, Frischer T, Barbato A. et al. Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. Eur Respir J 2012; 39: 1482-1491
- 104 Barbato A, Frischer T, Kuehni CE. et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. In: European Respiratory Journal. Eur Respir J 2009; 1264-1276
- 105 Lucas JS, Burgess A, Mitchison HM. et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 2014; 99: 850-856
- 106 El-Naggar AK, Chan JKC, Grandis JR et al. WHO Classification of Head
and Neck Tumours. 4th Edition Lyon: International Agency for
Research on Cancer; 2017
MissingFormLabel
- 107 Lombardi D, Tomenzoli D, Buttà L. et al. Limitations and complications of endoscopic surgery for treatment for sinonasal inverted papilloma: A reassessment after 212 cases. Head Neck 2011; 33: 1154-1161
- 108 Stange T, Schultz-Coulon HJ. Zum Chirurgischen Behandlungskonzept bei Invertierten Papillomen der Nase und Nasennebenhöhlen. HNO 2008; 56: 614-622
- 109 Christensen WN, Smith RRL. Schneiderian papillomas. A clinicopathologic study of 67 cases. Hum Pathol 1986; 17: 393-400
- 110 Stammberger H. New aspects in the genesis of inverted papillomas. Laryngol Rhinol Otol (Stuttg) 1983; 62: 249-24955
- 111 Leoncini G, Zanetti L. The papillomas of the sinonasal tract. A comprehensive review. Pathologica 2017; 109: 31-34
- 112 Sarkar FH, Visscher DW, Kintanar EB. et al. Sinonasal Schneiderian papillomas: human papillomavirus typing by polymerase chain reaction. Mod Pathol 1992; 5: 329-332
- 113 Barnes L. Schneiderian papillomas and nonsalivary glandular neoplasms of the head and neck. Mod Pathol 2002; 15: 279-297
- 114 Cunningham MJ, Brantley S, Barnes L. et al. Oncocytic Schneiderian papilloma in a young adult: A rare diagnosis. Otolaryngol Neck Surg 1987; 97: 47-51
- 115 Barnes L, Bedetti C. Oncocytic schneiderian papilloma: A reappraisal of cylindrical cell papilloma of the sinonasal tract. Hum Pathol 1984; 15: 344-351
- 116 Michaels L, Young M. Histogenesis of papillomas of the nose and paranasal sinuses. Arch Pathol Lab Med 1995; 119: 821-826
- 117 Lawson W, Patel ZM. The evolution of management for inverted papilloma: An analysis of 200 cases. Otolaryngol – Head Neck Surg 2009; 140: 330-335
- 118 Lund VJ, Stammberger H, Nicolai P. et al. European position paper on endoscopic management of tumours of the nose, paranasal sinuses and skull base. Rhinol Suppl 2010; 1-143
- 119 Maitra A, Baskin LB, Lee EL. Malignancies arising in oncocytic schneiderian papillomas: A report of 2 cases and review of the literature. Arch Pathol Lab Med 2001; 125: 1365-1367
- 120 Anari S, Carrie S. Sinonasal inverted papilloma: Narrative review. J Laryngol Otol 2010; 124: 705-715
- 121 Attlmayr B, Derbyshire SG, Kasbekar AV et al. Management of
inverted papilloma: Review. In: Journal of Laryngology and Otology.
Cambridge University Press; 2017: 284–289
MissingFormLabel
- 122 Heathcote KJ, Nair SB. The impact of modern techniques on the recurrence rate of inverted papilloma treated by endonasal surgery. Rhinology 2009; 47: 339-344
- 123 Diamantopoulos II, Jones NS, Lowe J. All nasal polyps need histological examination: An audit-based appraisal of clinical practice. J Laryngol Otol 2000; 114: 755-759
- 124 Lloyd GAS, Lloyd GAS. Epithelial Tumours. In: Diagnostic Imaging of the Nose and Paranasal Sinuses 1988; 95-110
- 125 Hildenbrand T, Weber R, Mertens J. et al. Surgery of Inverted Papilloma of the Maxillary Sinus via Translacrimal Approach – Long-Term Outcome and Literature Review. J Clin Med 2019; 8: 1873
- 126 Giotakis E, Knipping S, Kühnel T. et al. Unilateral diseases of the maxillary sinus. HNO 2020; 68: 623-636
- 127 Vrabec DP. The inverted schneiderian papilloma: A 25-year study. Laryngoscope 1994; 104: 582-605
- 128 Bielamowicz S, Calcaterra TC, Watson D. Inverting papilloma of the head and neck: The UCLA update. Otolaryngol – Head Neck Surg 1993; 109: 71-76
- 129 Kraft M, Simmen D, Kaufmann T. et al. Long-term results of endonasal sinus surgery in sinonasal papillomas. Laryngoscope 2003; 113: 1541-1547
- 130 Tanna N, Edwards JD, Aghdam H. et al. Transnasal endoscopic medial maxillectomy as the initial oncologic approach to sinonasal neoplasms: The anatomic basis. Arch Otolaryngol – Head Neck Surg 2007; 133: 1139-1142
- 131 Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: A meta-analysis. Otolaryngol – Head Neck Surg 2006; 134: 476-482
- 132 Veeresh M, Sudhakara M, Girish G. et al. Leiomyoma: A rare tumor in the head and neck and oral cavity: Report of 3 cases with review. J Oral Maxillofac Pathol 2013; 17: 281-287
- 133 Agaimy A, Michal M, Thompson LDR. et al. Angioleiomyoma of the Sinonasal Tract: Analysis of 16 Cases and Review of the Literature. Head Neck Pathol 2015; 9: 463-473
- 134 Heyman JNS, Jones LM, Hilton JM. et al. Angioleiomyoma of the inferior turbinate: A rare cause of isolated facial pain. Ann R Coll Surg Engl 2020; 102: E20-E22
- 135 Lau YW, Tharumalingam V, Tan TY. et al. Sinonasal angioleiomyoma. Med J Malaysia 2016; 71: 154-155
- 136 Mathieu T, Verbruggen A, Goovaerts G. et al. Vascular leiomyoma of the nasal cavity: case report and literature review. Eur Arch Oto-Rhino-Laryngology 2017; 274: 579-583
- 137 Poncet A, Dor L. Botryomycose humaine. Rev Chir 1897; 18: 996-1003
- 138 Hartzell M. Granuloma pyogenicum (botryomycosis of French authors). J Cutan Dis 1904; 22: 520-523
- 139 Mills SE, Cooper PH, Fechner RE. Lobular capillary hemangioma: The underlying lesion of pyogenic granuloma. A study of 73 cases from the oral and nasal mucous membranes. Am J Surg Pathol 1980; 4: 471-479
- 140 Fu Y-S, Perzin KH. Nonepithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx. A clinicopathologic study VI. Fibrous tissue tumors (fibroma, fibromatosis, fibrosarcoma). Cancer 1976; 37: 2912-2928
- 141 Thompson LDR, Fanburg-Smith JC. Update on Select Benign Mesenchymal and Meningothelial Sinonasal Tract Lesions. Head Neck Pathol 2016; 10: 95-108
- 142 Sammut SJ, Tomson N, Corrie P. Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med 2014; 371: 1265-1267
- 143 Smith SC, Patel RM, Lucas DR. et al. Sinonasal Lobular Capillary Hemangioma: A Clinicopathologic Study of 34 Cases Characterizing Potential for Local Recurrence. Head Neck Pathol 2013; 7: 129-134
- 144 Sunaryo PL, Svider PF, Husain Q. et al. Schwannomas of the sinonasal tract and anterior skull base: A systematic review of 94 cases. Am J Rhinol Allergy 2014; 28: 39-49
- 145 Shugar JMA, Son PM, Biller HF. et al. Peripheral nerve sheath tumors of the paranasal sinuses. Head Neck Surg 1981; 4: 72-76
- 146 Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994; 81: 362-373
- 147 Ansari I, Ansari A, Graison AA. et al. Head and Neck Schwannomas: A Surgical Challenge – A Series of 5 Cases. Case Rep Otolaryngol 2018; 2018: 1-10
- 148 Azani AB, Bishop JA, Thompson LDR. Sinonasal Tract Neurofibroma: A Clinicopathologic Series of 12 Cases with a Review of the Literature. Head Neck Pathol 2015; 9: 323-333
- 149 Annino DJ, Domanowski GF, Vaughan CW. A Rare Cause of Nasal Obstruction: A Solitary Neurofibroma. Otolaryngol Neck Surg 1991; 104: 484-488
- 150 Perzin KH, Panyu H, Wechter S. Nonepithelial tumors of the nasal cavity, paranasal sinuses and nasopharynx. A clinicopathologic study. XII: Schwann cell tumors (neurilemoma, neurofibroma, malignant schwannoma). Cancer 1982; 50: 2193-2202
- 151 Friedman CD, Costantino PD, Teitelbaum B. et al. Primary extracranial meningiomas of the head and neck. Laryngoscope 1990; 100: 41-48
- 152 Thompson LDR, Gyure KA. Extracranial sinonasal tract meningiomas: A clinicopathologic study of 30 cases with a review of the literature. Am J Surg Pathol 2000; 24: 640-650
- 153 Ho K-L. Primary meningioma of the nasal cavity and paranasal sinuses. Cancer 1980; 46: 1442-1447
- 154 Perry A, Scheithauer BW, Stafford SL. et al. „Malignancy“ in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999; 85: 2046-2056
- 155 Perry A, Stafford SL, Scheithauer BW. et al. Meningioma grading: An analysis of histologic parameters. Am J Surg Pathol 1997; 21: 1455-1465
- 156 Rushing EJ, Bouffard JP, McCall S. et al. Primary extracranial meningiomas: An analysis of 146 cases. Head Neck Pathol 2009; 3: 116-130
- 157 Mnejja M, Hammami B, Bougacha L. et al. Primary sinonasal meningioma. Eur Ann Otorhinolaryngol Head Neck Dis 2012; 129: 47-50
- 158
Wolf A,
Safran B,
Pock J.
et al. Surgical treatment of paranasal sinus osteomas: A single center experience
of 58
cases. Laryngoscope. 2019
MissingFormLabel
- 159 Atallah N, Jay MM. Osteomas of the paranasal sinuses. J Laryngol Otol 1981; 95: 291-304
- 160 Furlaneto EC, Rocha JRM, Heitz C. Osteoma of the zygomatic arch - Report of a case. Int J Oral Maxillofac Surg 2004; 33: 310-311
- 161 Summers LE, Mascott CR, Tompkins JR. et al. Frontal sinus osteoma associated with cerebral abscess formation: A case report. Surg Neurol 2001; 55: 235-239
- 162 Eller R, Sillers M. Common Fibro-osseous Lesions of the Paranasal Sinuses. Otolaryngol Clin North Am 2006; 39: 585-600
- 163 Schick B, Steigerwald C, El Tahan AER. et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology 2001; 39: 66-70
- 164 Schick B, Dlugaiczyk J. Benign tumors of the nasal cavity and paranasal sinuses. In: Rhinology and Facial Plastic Surgery 2009; 377-385
- 165 Buyuklu F, Akdogan MV, Ozer C. et al. Growth characteristics and clinical manifestations of the paranasal sinus osteomas. Otolaryngol - Head Neck Surg 2011; 145: 319-323
- 166 Golant A, Zeichner JA. Gardner Syndrome. StatPearls Publishing; 2014
- 167 Pinto RS, Simons A, Verma R. et al. Gardener-associated fibroma: An unusual cause of upper airway obstruction. BMJ Case Rep 2018; 2018
- 168 Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations
in hepatoblastoma. Pediatr Blood Cancer 2018; 65(4); doi:10.1002/
pbc.26892. Epub 2017 Dec 18. PMID: 29251405
MissingFormLabel
- 169 Kiessling P, Dowling E, Huang Y, Ho ML, Balakrishnan K, Weigel BJ,
Highsmith WE Jr, Niu Z, Schimmenti LA. Identification of aggressive
Gardner syndrome phenotype associated with a de novo APC variant,
c.4666dup. Cold Spring Harb Mol Case Stud 2019; 5(2): a003640.
doi:10.1101/mcs.a003640. PMID: 30696621; PMCID: PMC6549566
MissingFormLabel
- 170 Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis 2017; 23: 697-708
- 171 Erkmen K, Al-Mefty O, Adada B. Tumors of the skull base. In: Oncology of CNS Tumors Philadelphia: WB Saunders; 2010: 279-307
- 172 Heller AJ, DiNardo LJ, Massey D. Fibrous dysplasia, chondrosarcoma, and McCune-Albright syndrome. Am J Otolaryngol – Head Neck Med Surg 2001; 22: 297-301
- 173 Kos M, Luczak K, Godzinski J. et al. Treatment of monostotic fibrous dysplasia with pamidronate. J Cranio-Maxillofacial Surg 2004; 32: 10-15
- 174 Collins MT, Kushner H, Reynolds JC. et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res 2005; 20: 219-226
- 175 Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: Age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int 2008; 19: 57-63
- 176 Devogelaer JP. New uses of bisphosphonates: Osteogenesis imperfecta. Curr Opin Pharmacol 2002; 2: 748-753
- 177 Schreiber A, Villaret AB, Maroldi R. et al. Fibrous dysplasia of the sinonasal tract and adjacent skull base. Curr Opin Otolaryngol Head Neck Surg 2012; 20: 45-52
- 178 Amit M, Collins MT, FitzGibbon EJ. et al. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia – a meta-analysis. PLoS One 2011; 6
- 179 McCune D. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 1936; 52: 743-744
- 180 Leet AI, Chebli C, Kushner H. et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res 2004; 19: 571-577
- 181 Boyce AM, Brewer C, Deklotz TR. et al. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol – Head Neck Surg 2018; 144: 102-107
- 182 Deklotz TR, Kim HJ, Kelly M. et al. Sinonasal disease in polyostotic fibrous dysplasia and McCune-Albright Syndrome. Laryngoscope 2013; 123: 823-828
- 183 Boyce AM, Glover M, Kelly MH. et al. Optic neuropathy in McCune-Albright syndrome: Effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab 2013; 98
- 184 Plotkin H, Rauch F, Zeitlin L. et al. Effect of Pamidronate Treatment in Children with Polyostotic Fibrous Dysplasia of Bone. J Clin Endocrinol Metab 2003; 88: 4569-4575
- 185 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 2007; 4: 440-445
- 186 Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; (03) 12 PMID: 18489744 PMCID: PMC2459161
- 187 Feldman MD, Kelly M, Rao VM. et al. Fibrous dysplasia of the paranasal sinuses. Otolaryngol Neck Surg 1986; 95: 222-225
- 188 Ikeda K, Suzuki H, Oshima T. et al. Endonasal endoscopic management in fibrous dysplasia of the paranasal sinuses. Am J Otolaryngol – Head Neck Med Surg 1997; 18: 415-418
- 189 Uzun C, Adali MK, Koten M. et al. McCune-Albright syndrome with fibrous dysplasia of the paranasal sinuses. Rhinology 1999; 37: 122-124
- 190 Lee JS, Fitzgibbon E, Butman JA. et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 2002; 347: 1670-1676
- 191 Wenig BM, Heffner DK. Respiratory epithelial adenomatoid hamartomas of the sinonasal tract and nasopharynx: A clinicopathologic study of 31 cases. Ann Otol Rhinol Laryngol 1995; 104: 639-645
- 192 Delbrouck C, Fernandez Aguilar S, Choufani G. et al. Respiratory epithelial adenomatoid hamartoma associated with nasal polyposis. Am J Otolaryngol – Head Neck Med Surg 2004; 25: 282-284
- 193 Endo R, Matsuda H, Takahashi M. et al. Respiratory epithelial adenomatoid hamartoma in the nasal cavity. Acta Otolaryngol 2002; 122: 398-400
- 194 Ingram WF, Noone MC, Gillespie MB. Respiratory epithelial adenomatoid hamartoma: A case report. Ear, Nose Throat J 2006; 85: 190-192
- 195 Athre R, Ducic Y. Frontal sinus hamartomas. Am J Otolaryngol – Head Neck Med Surg 2005; 26: 419-421
- 196 Flavin R, Russell J, Phelan E. et al. Chondro-osseous respiratory epithelial adenomatoid hamartoma of the nasal cavity: A case report. Int J Pediatr Otorhinolaryngol 2005; 69: 87-91
- 197 Fitzhugh VA, Mirani N. Respiratory epithelial adenomatoid hamartoma: A review. Head Neck Pathol 2008; 2: 203-208
- 198 Braun H, Beham A, Stammberger H. Respiratorisches epitheliales adenomatoides hamartom der nasenhaupthöhle: Fallbericht und literaturübersicht. Laryngorhinootologie 2003; 82: 416-420
- 199 Himi Y, Yoshizaki T, Sato K. et al. Respiratory epithelial adenomatoid hamartoma of the maxillary sinus. J Laryngol Otol 2002; 116: 317-318
- 200 Safi C, Li C, Tabaee A. et al. Outcomes and imaging findings of respiratory epithelial adenomatoid hamartoma: a systematic review. Int Forum Allergy Rhinol 2019; 9: 674-680
- 201 Lima NB, Jankowski R, Georgel T. et al. Respiratory adenomatoid hamartoma must be suspected on CT-scan enlargement of the olfactory clefts. Rhinology 2006; 44: 264-269
- 202 Hawley KA, Ahmed M, Sindwani R. CT findings of sinonasal respiratory epithelial adenomatoid hamartoma: A closer look at the olfactory clefts. Am J Neuroradiol 2013; 34: 1086-1090
- 203 Metselaar RM, Stel HV, Van Der Baan S. Respiratory epithelial adenomatoid hamartoma in the nasopharynx. J Laryngol Otol 2005; 119: 476-478
- 204 Hawley KA, Pabon S, Hoschar AP. et al. The presentation and clinical significance of sinonasal respiratory epithelial adenomatoid hamartoma (REAH). Int Forum Allergy Rhinol 2013; 3: 248-253
- 205 Schafer DR, Thompson LDR, Smith BC. et al. Primary ameloblastoma of the sinonasal tract: A clinicopathologic study of 24 cases. Cancer 1998; 82: 667-674
- 206 Shahidi S, Bronoosh P, Daneshbod Y. Follicular ameloblastoma presenting as a sinonasal tumor. Iran Red Crescent Med J 2012; 14: 113-116
- 207 Hertog D, van der Waal I. Ameloblastoma of the jaws: A critical reappraisal based on a 40-years single institution experience. Oral Oncol 2010; 46: 61-64
- 208 Barrena BG, Phillips BJ, Moran KJ. et al. Sinonasal Ameloblastoma. Head Neck Pathol 2019; 13: 247-250
- 209
Golbin DA, Ektova AP, Demin MO, Lasunin N, Cherekaev VA. Nasal
Chondromesenchymal Hamartoma with Skull Base and Orbital
Involvement: Case Presentation. Cureus 2018; 10(6): e2892.
doi:10.7759/cureus.2892. PMID: 30167348; PMCID: PMC6112910
MissingFormLabel
- 210 McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: An upper respiratory tract analogue of the chest wall mesenchymal hamartoma. Am J Surg Pathol 1998; 22: 425-433
- 211 Ozolek JA, Carrau R, Barnes EL. et al. Nasal chondromesenchymal hamartoma in older children and adults: Series and immunohistochemical analysis. Arch Pathol Lab Med 2005; 129: 1444-1450
- 212 Chan TY. World Health Organization classification of tumours: Pathology & genetics of tumours of the urinary system and male genital organs. Urology 2005; 65: 214-215
- 213 Seibert RW, Seibert JJ, Jimenez JF. et al. Nasopharyngeal brain heterotopia – a cause of upper airway obstruction in infancy. Laryngoscope 1984; 94: 818-819
- 214 Kanjanawasee D, Chaowanapanja P, Keelawat S. et al. Sphenoid Sinus Cholesteatoma – Complications and Skull Base Osteomyelitis: Case Report and Review of Literature. Clin Med Insights Case Reports 2019; 12
- 215 Hansen S, Sørensen CH, Stage J. et al. Massive cholesteatoma of the frontal sinus: Case report and review of the literature. Auris Nasus Larynx 2007; 34: 387-392
- 216
Gey A, Plontke SK, Scheller C, Kösling S, Fathke C, Glien A. Seltene
Diagnose einer knochendestruierenden Läsion der Keilbeinhöhle
[Rare diagnosis of a bone-destructive lesion of the sphenoid sinus].
HNO. 2020 Dec 14. German. doi:10.1007/s00106-020-00974-2.
Epub ahead of print. PMID: 33315128
MissingFormLabel
- 217 Ansa B, Goodman M, Ward K. et al. Paranasal sinus squamous cell carcinoma incidence and survival based on surveillance, epidemiology, and end results data, 1973 to 2009. Cancer 2013; 119: 2602-2610
- 218 Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2012; 34: 877-885
- 219 Sanghvi S, Khan MN, Patel NR. et al. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2014; 124: 76-83
- 220 Youlden DR, Cramb SM, Peters S. et al. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol 2013; 37: 770-779
- 221 Hayes RB, Kardaun JWPF, De Bruyn A. Tobacco use and sinonasal cancer: A case-control study. Br J Cancer 1987; 56: 843-846
- 222 Brinton LA, Blot WJ, Becker JA. et al. A case-control study of cancers of the nasal cavity and paranasal sinuses. Am J Epidemiol 1984; 119: 896-906
- 223 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian Papilloma (Malignant Transformation): A Clinicopathologic and Immunophenotypic Study of 20 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2014; 8: 269-286
- 224 Dulguerov P, Jacobsen M, Allal A. et al. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer 2001; 92: 3012-3029
- 225 Thorup C, Sebbesen L, Danø H. et al. Carcinoma of the nasal cavity and paranasal sinuses in Denmark 1995-2004. Acta Oncol (Madr) 2010; 49: 389-394
- 226 El-Mofty SK, Lu DW. Prevalence of high-risk human papillomavirus DNA in nonkeratinizing (cylindrical cell) carcinoma of the sinonasal tract: A distinct clinicopathologic and molecular disease entity. Am J Surg Pathol 2005; 29: 1367-1372
- 227 Bishop JA, Guo TW, Smith DF. et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am J Surg Pathol 2013; 37: 185-192
- 228 Osborn DA. Nature and behavior of transitional tumors in the upper respiratory tract. Cancer 1970; 25: 50-60
- 229 Robin PE, Powell DJ, Stansbie JM. Carcinoma of the nasal cavity and paranasal sinuses: incidence and presentation of different histological types. Clin Otolaryngol Allied Sci 1979; 4: 431-456
- 230 Larque AB, Hakim S, Ordi J. et al. High-risk human papillomavirus is transcriptionally active in a subset of sinonasal squamous cell carcinomas. Mod Pathol 2014; 27: 343-351
- 231 Mirghani H, Hartl D, Mortuaire G. et al. Nodal recurrence of sinonasal cancer: Does the risk of cervical relapse justify a prophylactic neck treatment?. Oral Oncol 2013; 49: 374-380
- 232 Castelnau-Marchand P, Levy A, Moya-Plana A. et al. Sinunasales Plattenepithelkarzinom ohne klinische Lymphknotenbeteiligung: Welches Halsmanagement ist das Beste?. Strahlentherapie und Onkol 2016; 192: 537-544
- 233 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian
Papilloma (Malignant Transformation): A Clinicopathologic and
Immunophenotypic Study of 20 Cases Combined with a Comprehensive
Review of the Literature. Head Neck Pathol 2014; 8: 269–286
MissingFormLabel
- 234 Furuta A, Kudo M, Kanai KI. et al. Typical carcinoid tumor arising in the nose and paranasal sinuses – Case report. Auris Nasus Larynx 2010; 37: 381-385
- 235 Guan M, Li Y, Shi ZG. et al. Sarcomatoid carcinoma involving the nasal cavity and paranasal sinus: A rare and highly progressive tumor. Int J Clin Exp Pathol 2014; 7: 4489-4492
- 236 Thompson LDR, Wieneke JA, Miettinen M. et al. Spindle cell (sarcomatoid) carcinomas of the larynx: A clinicopathologic study of 187 cases. Am J Surg Pathol 2002; 26: 153-170
- 237 Lewis JE, Olsen KD, Sebo TJ. Spindle cell carcinoma of the larynx: Review of 26 cases including DNA content and immunohistochemistry. Hum Pathol 1997; 28: 664-673
- 238 Jeng YM, Sung MT, Fang CL. et al. Sinonasal undifferentiated carcinoma and nasopharyngeal-type undifferentiated carcinoma: Two clinically, biologically, and histopathologically distinct entities. Am J Surg Pathol 2002; 26: 371-376
- 239 Leung SuetYi, Yuen SiuTsan, Chung LapPing. et al. Epstein-Barr virus is present in a wide histological spectrum of sinonasal carcinomas. Am J Surg Pathol 1995; 19: 994-1001
- 240
Takakura H, Tachino H, Fujisaka M, Nakajima T, Yamagishi K, Ishida M,
Shojaku H. Lymphoepithelial carcinoma of the maxillary sinus: A case
report and review of the literature. Medicine (Baltimore) 2018;
97(28): e11371. doi:10.1097/MD.0000000000011371. PMID:
29995775; PMCID: PMC6076030
MissingFormLabel
- 241 Frierson HF, Mills SE, Fechner RE. et al. Sinonasal undifferentiated carcinoma. An aggressive neoplasm derived from Schneiderian epithelium and distinct from olfactory neuroblastoma. Am J Surg Pathol 1986; 10: 771-779
- 242 Cerilli LA, Holst VA, Brandwein MS. et al. Sinonasal undifferentiated carcinoma: Immunohistochemical profile and lack of EBV association. Am J Surg Pathol 2001; 25: 156-163
- 243
Abdelmeguid AS, Bell D, Hanna EY. Sinonasal Undifferentiated
Carcinoma. Curr Oncol Rep 2019; 21(3): 26. doi:10.1007/s11912-
019-0776-4. PMID: 30806835
MissingFormLabel
- 244 Musy PY, Reibel JF, Levine PA. Sinonasal undifferentiated carcinoma; The search for a better outcome. Laryngoscope 2002; 112: 1450-1455
- 245 Schröck A, Göke F, van Bremen T. et al. Maligne erkrankungen des sinunasaltrakts. Eine monoinstitutionelle erfahrung von 1996 bis 2010. HNO 2012; 60: 1041-1046
- 246 Rosenthal DI, Barker JL, El-Naggar AK. et al. Sinonasal malignancies with neuroendocrine differentiation: Patterns of failure according to histologic phenotype. Cancer 2004; 101: 2567-2573
- 247 Bell D, Hanna EY. Sinonasal undifferentiated carcinoma: Morphological heterogeneity, diagnosis, management and biological markers. Expert Rev Anticancer Ther 2013; 13: 285-296
- 248 Chambers KJ, Lehmann AE, Remenschneider A. et al. Incidence and survival patterns of sinonasal undifferentiated carcinoma in the United States. J Neurol Surgery, Part B Skull Base 2015; 76: 94-100
- 249 Kuan EC, Arshi A, Mallen-St Clair J, Tajudeen BA, Abemayor E, St John
MA. Significance of Tumor Stage in Sinonasal Undifferentiated
Carcinoma Survival: A Population-Based Analysis. Otolaryngol Head
Neck Surg 2016; 154(4): 667–673. doi:10.1177/01945998
16629649. Epub 2016 Feb 23. PMID: 26908559
MissingFormLabel
- 250 Takahashi Y, Lee J, Pickering C. et al. Human epidermal growth factor receptor 2/neu as a novel therapeutic target in sinonasal undifferentiated carcinoma. Head Neck 2016; 38: E1926-E1934
- 251 Sanchez-Casis G, Devine KD, Weiland LH. Nasal adenocarcinomas that closely simulate colonic carcinomas. Cancer 1971; 28: 714-720
- 252 Barnes L. Intestinal-type adenocarcinoma of the nasal cavity and paranasal sinuses. Am J Surg Pathol 1986; 10: 192-202
- 253 Szablewski V, Solassol J, Poizat F. et al. EGFR expression and KRAS and BRAF mutational status in intestinal-type sinonasal adenocarcinoma. Int J Mol Sci 2013; 14: 5170-5181
- 254 Saber AT, Nielsen LR, Dictor M. et al. K-ras mutations in sinonasal adenocarcinomas in patients occupationally exposed to wood or leather dust. Cancer Lett 1998; 126: 59-65
- 255 Franchi A, Innocenti DRD, Palomba A. et al. Low prevalence of K-RAS, EGF-R and BRAF mutations in sinonasal adenocarcinomas. Implications for anti-EGFR treatments. Pathol Oncol Res 2014; 20: 571-579
- 256 Projetti F, Durand K, Chaunavel A. et al. Epidermal growth factor receptor expression and KRAS and BRAF mutations: Study of 39 sinonasal intestinal-type adenocarcinomas. Hum Pathol 2013; 44: 2116-2125
- 257 Ferrari M, Bossi P, Mattavelli D, Ardighieri L, Nicolai P. Management of
sinonasal adenocarcinomas with anterior skull base extension. J
Neurooncol 2020;150(3): 405–417. doi:10.1007/s11060-019-
03385-8. Epub 2020 Jan 3. PMID: 31897925
MissingFormLabel
- 258 Hoffmann TK, El Hindy N, Müller OM. et al. Vascularised local and free flaps in anterior skull base reconstruction. Eur Arch Oto-Rhino-Laryngology 2013; 270: 899-907
- 259 Hoeben A, van de Winkel L, Hoebers F. et al. Intestinal-type sinonasal adenocarcinomas: The road to molecular diagnosis and personalized treatment. Head Neck 2016; 38: 1564-1570
- 260 Turri-Zanoni M, Battaglia P, Lambertoni A. et al. Treatment strategies for primary early-stage sinonasal adenocarcinoma: A retrospective bi-institutional case-control study. J Surg Oncol 2015; 112: 561-567
- 261 Licitra L, Locati LD, Cavina R. et al. Primary chemotherapy followed by anterior craniofacial resection and radiotherapy for paranasal cancer. Ann Oncol 2003; 14: 367-372
- 262 Jo VY, Mills SE, Cathro HP. et al. Low-grade sinonasal adenocarcinomas: The association with and distinction from respiratory epithelial adenomatoid hamartomas and other glandular lesions. Am J Surg Pathol 2009; 33: 401-408
- 263 Neto AG, Pineda-Daboin K, Luna MA. Sinonasal tract seromucous adenocarcinomas: A report of 12 cases. Ann Diagn Pathol 2003; 7: 154-159
- 264 Heffner DK, Hyams VJ, Hauck KW. et al. Low-grade adenocarcinoma of the nasal cavity and paranasal sinuses. Cancer 1982; 50: 312-322
- 265 Stelow EB, Jo VY, Mills SE. et al. A histologic and immunohistochemical study describing the diversity of tumors classified as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol 2011; 35: 971-980
- 266 Bignami M, Lepera D, Volpi L. et al. Sinonasal Non-Intestinal-Type Adenocarcinoma: A Retrospective Review of 22 Patients. World Neurosurg 2018; 120: e962-e969
- 267 Shay A, Ganti A, Raman A. et al. Survival in low-grade and high-grade sinonasal adenocarcinoma: A national cancer database analysis. Laryngoscope 2020; 130: E1-E10
- 268 Shen T, Shi Q, Velosa C. et al. Sinonasal renal cell-like adenocarcinomas: Robust carbonic anhydrase expression. Hum Pathol 2015; 46: 1598-1606
- 269 Heffner DK. Sinonasal and laryngeal salivary gland lesions. In: Surgical Pathology of the Salivary Glands. Philadelphia: WB Saunders; 1991: 544-559
- 270
Akbaba S, Ahmed D, Mock A, Held T, Bahadir S, Lang K, Syed M,
Hoerner-Rieber J, Forster T, Federspil P, Herfarth K, Plinkert P, Debus
J, Adeberg S. Treatment Outcome of 227 Patients with Sinonasal Adenoid
Cystic Carcinoma (ACC) after Intensity Modulated Radiotherapy
and Active Raster-Scanning Carbon Ion Boost: A 10-Year Single-Center
Experience. Cancers (Basel) 2019; 111(11): 1705–417.
doi:10.3390/cancers11111705. PMID: 31683896; PMCID:
PMC6895865
MissingFormLabel
- 271 Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study of 242 cases. Am J Surg 1974; 128: 512-520
- 272 Spiro RH. Salivary neoplasms: Overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8: 177-184
- 273 Michel G, Joubert M, Delemazure AS. et al. Adenoid cystic carcinoma of the paranasal sinuses: Retrospective series and review of the literature. Eur Ann Otorhinolaryngol Head Neck Dis 2013; 130: 257-262
- 274 Kim GE, Park HC, Keum KC. et al. Adenoid cystic carcinoma of the maxillary antrum. Am J Otolaryngol – Head Neck Med Surg 1999; 20: 77-84
- 275 Chen AM, Bucci MK, Weinberg V. et al. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: Prognostic features of recurrence. Int J Radiat Oncol Biol Phys 2006; 66: 152-159
- 276 Terhaard CHJ, Lubsen H, Rasch CRN. et al. The role of radiotherapy in the treatment of malignant salivary gland tumors. Int J Radiat Oncol Biol Phys 2005; 61: 103-111
- 277 Garden AS, Weber RS, Ang KK. et al. Postoperative radiation therapy for malignant tumors of minor salivary glands. Outcome and patterns of failure. Cancer 1994; 73: 2563-2569
- 278 Chen AM, Granchi PJ, Garcia J. et al. Local-regional recurrence after surgery without postoperative irradiation for carcinomas of the major salivary glands: Implications for adjuvant therapy. Int J Radiat Oncol Biol Phys 2007; 67: 982-987
- 279 Mendenhall WM, Morris CG, Amdur RJ. et al. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck 2004; 26: 154-162
- 280 Terhaard CHJ, Lubsen H, Van Der Tweel I. et al. Salivary gland carcinoma: Independent prognostic factors for locoregional control, distant metastases, and overall survival: Results of the Dutch Head and Neck Oncology Cooperative Group. Head Neck 2004; 26: 681-693
- 281 Katz TS, Mendenhall WM, Morris CG. et al. Malignant tumors of the nasal cavity and paranasal sinuses. Head Neck 2002; 24: 821-829
- 282 Laramore GE, Krall JM, Griffin TW. et al. Neutron versus photon irradiation for unresectable salivary gland tumors: Final report of an RTOG-MRC randomized clinical trial. Int J Radiat Oncol Biol Phys 1993; 27: 235-240
- 283 Douglas JG, Koh WJ, Austin-Seymour M. et al. Treatment of salivary gland neoplasms with fast neutron radiotherapy. Arch Otolaryngol - Head Neck Surg 2003; 129: 944-948
- 284 Stannard C, Vernimmen F, Carrara H. et al. Malignant salivary gland tumours: Can fast neutron therapy results point the way to carbon ion therapy?. Radiother Oncol 2013; 109: 262-268
- 285 Huber PE, Debus J, Latz D. et al. Radiotherapy for advanced adenoid cystic carcinoma: Neutrons, photons or mixed beam?. Radiother Oncol 2001; 59: 161-167
- 286 Pommier P, Liebsch NJ, Deschler DG. et al. Proton beam radiation therapy for skull base adenoid cystic carcinoma. Arch Otolaryngol - Head Neck Surg 2006; 132: 1242-1249
- 287 Patel NR, Sanghvi S, Khan MN. et al. Demografic trends and disease-specific survival in salivary acinic cell carcinoma: An analysis of 1129 cases. Laryngoscope 2014; 124: 172-178
- 288 Haerle SK, Gullane PJ, Witterick IJ. et al. Sinonasal Carcinomas. Epidemiology, Pathology, and Management. Neurosurg Clin N Am 2013; 24: 39-49
- 289 Neto AG, Pineda-Daboin K, Spencer ML. et al. Sinonasal acinic cell carcinoma: A clinicopathologic study of four cases. Head Neck 2005; 27: 603-607
- 290 Wong A, Leong JL, Ho B. Primary acinic cell carcinoma of the ethmoid sinus. Ear, Nose Throat J 2010; 89
- 291 Biron VL, Lentsch EJ, Gerry DR. et al. Case-control analysis of survival outcomes in sinonasal acinic cell carcinoma. Int Forum Allergy Rhinol 2014; 4: 507-511
- 292 Ellis MA, Graboyes EM, Day TA. et al. Prognostic factors and occult nodal disease in mucoepidermoid carcinoma of the oral cavity and oropharynx: An analysis of the National Cancer Database. Oral Oncol 2017; 72: 174-178
- 293
Auger SR,
Patel T,
Ganti A.
et al. Effect of margin status and pathological grade in treatment of sinonasal
mucoepidermoid carcinoma. Laryngoscope. 2020
MissingFormLabel
- 294 Bhattacharyya N. Survival and staging characteristics for non-squamous cell malignancies of the maxillary sinus. Arch Otolaryngol - Head Neck Surg 2003; 129: 334-337
- 295 Wolfish EB, Nelson BL, Thompson LDR. Sinonasal Tract Mucoepidermoid Carcinoma: A Clinicopathologic and Immunophenotypic Study of 19 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2012; 6: 191-207
- 296 Lee YS, Ha SM, Paik SW. et al. Epithelial-myoepithelial carcinoma originating from a minor salivary gland in the nasal septum: A case report and literature review. Medicine (Baltimore) 2020; 99: e19072
- 297 Gore MR. Epithelial-myoepithelial carcinoma: A population-based survival analysis. BMC Ear, Nose Throat Disord 2018; 18
- 298 Morresi-Hauf AT, Reu S, Fertl A. Epithelial-myoepitheliales Karzinom der Trachea: Fallbericht und Literaturübersicht. Pathologe 2013; 34: 56-64
- 299 Roh JL, Chang HR, Choi SH. et al. Clinical utility of 18F-FDG PET for patients with salivary gland malignancies. J Nucl Med 2007; 48: 240-246
- 300 Kim CH, Jeong JS, Kim SR. et al. Endobronchial epithelial-myoepithelial carcinoma of the lung. Thorax 2018; 73: 593-594
- 301 Kim SH, Park SE, Bae HG. et al. Epithelial-myoepithelial carcinoma of the nasopharynx: A case report and review of the literature. Oncol Lett 2015; 10: 927-930
- 302 Fried D, Zanation AM, Huang B. et al. Management of nonesthesioneuroblastoma sinonasal malignancies with neuroendocrine differentiation. Laryngoscope 2012; 122: 2210-2215
- 303 Hong SL, Kim SD, Roh HJ. et al. The sphenoid sinus: An unusual presentation of a typical carcinoid tumor. J Craniofac Surg 2014; 25: e483-e485
- 304 Konukiewitz B, Agaimy A, Weichert W. et al. Neuroendokrine Neoplasien der Kopf-Hals-Region. Pathologe 2018; 39: 27-34
- 305 Ferlito A, Silver CE, Bradford CR. et al. Neuroendocrine neoplasms of the larynx: An overview. Head Neck 2009; 31: 1634-1646
- 306 Lee DH, Cho HH, Cho YB. Typical carcinoid tumor of the nasal cavity. Auris Nasus Larynx 2007; 34: 537-539
- 307 Patel TD, Carniol ET, Vázquez A. et al. Sinonasal fibrosarcoma: Analysis of the Surveillance, Epidemiology, and End Results database. Int Forum Allergy Rhinol 2016; 6: 201-205
- 308 Lartigau E, Lusinchi A, Schwaab G. Sarcomas of nasal cavity and paranasal sinuses: Chondrosarcoma, osteosarcoma and fibrosarcoma. J Laryngol Otol 1994; 108: 947-953
- 309 Koshy M, Rich SE, Mohiuddin MM. Improved Survival With Radiation Therapy in High-Grade Soft Tissue Sarcomas of the Extremities: A SEER Analysis. Int J Radiat Oncol Biol Phys 2010; 77: 203-209
- 310 Wang CP, Chang YL, Ting LL. et al. Malignant fibrous histiocytoma of the sinonasal tract. Head Neck 2009; 31: 85-93
- 311 Szablewski V, Neuville A, Terrier P. et al. Adult sinonasal soft tissue sarcoma: Analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 2015; 125: 615-623
- 312 Gerrand CH, Bell RS, Wunder JS. et al. The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer 2003; 97: 485-492
- 313 Kwok MMK, Lee S, Hosking P. Leiomyosarcoma: A rare sinonasal malignancy. BMJ Case Rep 2018; 2018
- 314 Tanaka H, Westesson PL, Wilbur DC. Leiomyosarcoma of the maxillary sinus: CT and MRI findings. Br J Radiol 1998; 71: 221-224
- 315 Dobben GD. Leiomyosarcoma of the Nasopharynx. AMA Arch Otolaryngol 1958; 68: 211-213
- 316 Sanghvi S, Misra P, Patel NR. et al. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol - Head Neck Med Surg 2013; 34: 682-689
- 317 Wurm J, Constantinidis J, Grabenbauer GG. et al. Rhabdomyosarcomas of the nose and paranasal sinuses: Treatment results in 15 cases. Otolaryngol - Head Neck Surg 2005; 133: 42-50
- 318 Narula AA, Vallis MP, El-Silimy OE. et al. Radiation induced angiosarcomas of the nasopharynx. Eur J Surg Oncol 1986; 12: 147-152
- 319 Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: A study of forty-four cases. Cancer 1981; 48: 1907-1921
- 320 Williamson IG, Ramsden RT. Angiosarcoma of maxillary antrum—association with vinyL chloride exposure. J Laryngol Otol 1988; 102: 464-467
- 321 Nelson BL, Thompson LDR. Sinonasal tract angiosarcoma: A clinicopathologic and immunophenotypic study of 10 cases with a review of the literature. Head Neck Pathol 2007; 1: 1-12
- 322 Fukushima K, Dejima K, Koike S. et al. A case of angiosarcoma of the nasal cavity successfully treated with recombinant interleukin-2. Otolaryngol - Head Neck Surg 2006; 134: 886-887
- 323 Treviño-González JL, Santos-Lartigue R, González-Andrade B. et al. Angiosarcoma of the nasal cavity: A case report. Cases J 2009; 2
- 324 Es-Sbissi F, Nitassi S, Boulaadas M. et al. Sinonasal angiosarcoma. Eur Ann Otorhinolaryngol Head Neck Dis 2015; 132: 161-163
- 325 Agaimy A, Hartmann A. Kopf-Hals-Tumoren: Neues aus der WHO-Klassifikation 2017. Pathologe 2018; 39
- 326 Agaimy A, Haller F, Hartmann A. Sinunasale Tumoren: Neues aus der WHO mit besonderem Fokus auf neue mesenchymale Entitäten. Pathologe 2018; 39: 18-26
- 327 Lewis JT, Oliveira AM, Nascimento AG. et al. Low-grade sinonasal sarcoma with neural and myogenic features: A clinicopathologic analysis of 28 cases. Am J Surg Pathol 2012; 36: 517-525
- 328 Rooper LM, Huang SC, Antonescu CR. et al. Biphenotypic sinonasal sarcoma: An expanded immunoprofile including consistent nuclear β-catenin positivity and absence of SOX10 expression. Hum Pathol 2016; 55: 44-50
- 329 Carter CS, East EG, McHugh JB. Biphenotypic sinonasal sarcoma: A review and update. In: Archives of Pathology and Laboratory Medicine. College of American Pathologists 2018; 1196-1201
- 330 Fritchie KJ, Jin L, Wang X. et al. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology 2016; 69: 930-936
- 331 Ducatman BS, Scheithauer BW, Piepgras DG. et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57: 2006-2021
- 332 Rodriguez FJ, Folpe AL, Giannini C. et al. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012; 123: 295-319
- 333 James AW, Shurell E, Singh A. et al. Malignant Peripheral Nerve Sheath Tumor. Surg Oncol Clin N Am 2016; 25: 789-802
- 334 Bradtmöller M, Hartmann C, Zietsch J. et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One 2012; 7: e47595
- 335 Kar M, Suryanarayana Deo SV, Shukla NK. et al. Malignant peripheral nerve sheath tumors (MPNST) - Clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol 2006; 4: 55
- 336 Jo VY, Fletcher CDM. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014; 46: 95-104
- 337 Van De Rijn M, Barr FG, Xiong QBin. et al. Radiation-associated synovial sarcoma. Hum Pathol 1997; 28: 1325-1328
- 338 Deraedt K, Debiec-Rychter M, Sciot R. Radiation-associated synovial sarcoma of the lung following radiotherapy for pulmonary metastasis of Wilms’ tumour [7]. Histopathology 2006; 48: 473-475
- 339 Egger JF, Coindre JM, Benhattar J. et al. Radiation-associated synovial sarcoma: Clinicopathologic and molecular analysis of two cases. Mod Pathol 2002; 15: 998-1004
- 340 Sherman KL, Wayne JD, Chung J. et al. Assessment of multimodality therapy use for extremity sarcoma in the United States. J Surg Oncol 2014; 109: 395-404
- 341 Vlenterie M, Jones RL, Van Der Graaf WTA. Synovial sarcoma diagnosis and management in the era of targeted therapies. Curr Opin Oncol 2015; 27: 316-322
- 342 De Bree E, Zoras O, Hunt JL. et al. Desmoid tumors of the head and neck: A therapeutic challenge. Head Neck 2014; 36: 1517-1526
- 343 Orphanet: Desmoid type fibromatosis. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=8665&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=873&Disease(s)/groupofdiseases=Desmoid-type-fibromatosis&title=Desmoid-type-fibromatosis&search=Dise
MissingFormLabel
- 344 Colombo C, Foo WC, Whiting D. et al. FAP-related desmoid tumors: A series of 44 patients evaluated in a cancer referral center. Histol Histopathol 2012; 27: 641-649
- 345 Coffin CM, Hornick JL, Zhou H. et al. Gardner fibroma: A clinicopathologic and immunohistochemical analysis of 45 patients with 57 fibromas. Am J Surg Pathol 2007; 31: 410-416
- 346 Emori M, Kaya M, Mitsuhashi T. et al. Desmoid tumor-associated pain is dependent on mast cell expression of cyclooxygenase-2. Diagn Pathol 2014; 9
- 347 Kasper B, Baumgarten C, Garcia J. et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol 2017; 28: 2399-2408
- 348 Bonvalot S, Eldweny H, Haddad V. et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 2008; 34: 462-468
- 349 Penel N, Chibon F, Salas S. Adult desmoid tumors: Biology, management and ongoing trials. Curr Opin Oncol 2017; 29: 268-274
- 350 Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: A clinicopathological and immunohistochemical analysis of 46 cases. Pathology 2010; 42: 147-150
- 351 Granter SR, Badizadegan K, Fletcher CDM. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: A spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998; 22: 513-525
- 352 Asimakopoulos P, Syed MI, Andrews T. et al. Sinonasal glomangiopericytoma: Is anything new?. Ear, Nose Throat J 2016; 95: E1
- 353 Gengler C, Guillou L. Solitary fibrous tumour and haemangiopericytoma: Evolution of a concept. Histopathology 2006; 48: 63-74
- 354 Thompson LDR, Miettinen M, Wenig BM. Sinonasal-type hemangiopericytoma: A clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol 2003; 27: 737-749
- 355 Catalano PJ, Brandwein M, Shah DK. et al. Sinonasal hemangiopericytomas: A clinicopathologic and immunohistochemical study of seven cases. Head Neck 1996; 18: 42-53
- 356 Billings KR, Fu YS, Calcaterra TC. et al. Hemangiopericytoma of the head and neck. Am J Otolaryngol - Head Neck Med Surg 2000; 21: 238-243
- 357 Carew JF, Singh B, Kraus DH. Hemangiopericytoma of the head and neck. Laryngoscope 1999; 109: 1409-1411
- 358 Weber W, Henkes H, Metz KA. et al. Haemangiopericytoma of the nasal cavity. Neuroradiology 2001; 43: 183-186
- 359 Witkin GB, Rosai J. Solitary fibrous tumor of the upper respiratory tract: A report of six cases. Am J Surg Pathol 1991; 15: 842-848
- 360 Batsakis JG, El-Naggar AK, Hybels RD. Pathology consultation solitary fibrous tumor. Ann Otol Rhinol Laryngol 1993; 102: 74-76
- 361 Künzel J, Hainz M, Ziebart T. et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1589-1598
- 362
Demicco EG,
Park MS,
Araujo DM.
et al. Solitary fibrous tumor: A clinicopathological study of 110 cases and proposed
risk assessment model. Mod Pathol 2012; 25: 1298-1306
MissingFormLabel
- 363 Xue Y, Chai G, Xiao F. et al. Post-operative radiotherapy for the treatment of malignant solitary fibrous tumor of the nasal and paranasal area. Jpn J Clin Oncol 2014; 44: 926-931
- 364 Piccaluga PP, De Falco G, Kustagi M. et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011; 117: 3596-3608
- 365 Chi AC, Weathers DR, Folpe AL. et al. Epithelioid hemangioendothelioma of the oral cavity: Report of two cases and review of the literature. Oral Surgery, Oral Med Oral Pathol Oral Radiol Endodontology 2005; 100: 717-724
- 366 Errani C, Zhang L, Sung YS. et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosom Cancer 2011; 50: 644-653
- 367 Flucke U, Vogels RJC, de Saint Aubain Somerhausen N. et al. Epithelioid Hemangioendothelioma: Clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol 2014; 9
- 368 Bruder E, Alaggio R, Kozakewich HPW. et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol 2012; 15: 26-61
- 369 Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma a vascular tumor often mistaken for a carcinoma. Cancer 1982; 50: 970-981
- 370 Mentzel T, Beham A, Calonje E. et al. Epithelioid hemangioendothelioma of skin and soft tissues: Clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997; 21: 363-374
- 371 Deyrup AT, Tighiouart M, Montag AG. et al. Epithelioid hemangioendothelioma of soft tissue: A proposal for risk stratification based on 49 cases. Am J Surg Pathol 2008; 32: 924-927
- 372 Beger M, Antoni C, Haas S. et al. Epitheloides Hämangioendotheliom- drei Patienten, drei Therapieoptionen. Z Gastroenterol 2006; 44: A5_04
- 373 Wong BLK, Lee VNY, Tikka T. et al. Kaposiform haemangioendothelioma of the head and neck. Crit Rev Oncol Hematol 2016; 104: 156-168
- 374 Sun ZJ, Zhang L, Zhang WF. et al. Kaposiform hemangioendothelioma involving the neck. Oral Oncol Extra 2006; 42: 60-65
- 375 Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis 2013; 8: 95
- 376 Pongpruttipan T, Sukpanichnant S, Assanasen T. et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and αβ, γδ, and αβ/γδ T-cell origin: A comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol 2012; 36: 481-499
- 377 Jhuang JY, Chang ST, Weng SF. et al. Extranodal natural killer/T-cell lymphoma, nasal type in Taiwan: A relatively higher frequency of T-cell lineage and poor survival for extranasal tumors. Hum Pathol 2015; 46: 313-321
- 378 De Campos-Lima PO, Gavioli R, Zhang QJ. et al. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11 + population. Science (80-) 1993; 260: 98-100
- 379 Lima PODC, Levitsky V, Brooks J. et al. T cell responses and virus evolution: Loss of hla all-restricted ctl epitopes in epstein-barr virus isolates from highly all-positive populations by selective mutation of anchor residues. J Exp Med 1994; 179: 1297-1305
- 381 Kim SJ, Choi JY, Hyun SH. et al. Risk stratification on the basis of Deauville score on PET-CT and the presence of Epstein-Barr virus DNA after completion of primary treatment for extranodal natural killer/T-cell lymphoma, nasal type: A multicentre, retrospective analysis. Lancet Haematol 2015; 2: e66-e74
- 381 Tse E, Kwong YL. How i treat NK/T-cell lymphomas. Blood 2013; 121: 4997-5005
- 382 Kwong YL, Kim WS, Lim ST. et al. SMILE for natural killer/T-cell lymphoma: Analysis of safety and efficacy from the Asia Lymphoma Study Group. Blood 2012; 120: 2973-2980
- 383 Alexiou C, Kau RJ, Dietzfelbinger H. et al. Extramedullary plasmacytoma: Tumor occurrence and therapeutic concepts. Cancer 1999; 85: 2305-2314
- 384 Miller FR, Lavertu P, Wanamaker JR. et al. Plasmacytomas of the head and neck. Otolaryngol - Head Neck Surg 1998; 119: 614-618
- 385 Patel TD, Vázquez A, Choudhary MM. et al. Sinonasal extramedullary plasmacytoma: A population-based incidence and survival analysis. Int Forum Allergy Rhinol 2015; 5: 862-869
- 386 Soutar R, Lucraft H, Jackson G. et al. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Br J Haematol 2004; 124: 717-726
- 387 Lorsbach RB, Hsi ED, Dogan A. et al. Plasma cell myeloma and related neoplasms. In: American Journal of Clinical Pathology 2011; 168-182
- 388 Susnerwala SS, Shanks JH, Banerjee SS. et al. Extramedullary plasmacytoma of the head and neck region: Clinicopathological correlation in 25 cases. Br J Cancer 1997; 75: 921-927
- 389 Hussong JW, Perkins SL, Schnitzer B. et al. Extramedullary plasmacytoma: A form of marginal zone cell lymphoma?. Am J Clin Pathol 1999; 111: 111-116
- 390 Boll M, Parkins E, O’Connor SJM. et al. Extramedullary plasmacytoma are characterized by a „myeloma-like“ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol 2010; 151: 525-527
- 391 Galieni P, Cavo M, Pulsoni A. et al. Clinical outcome of extramedullary plasmacytoma. Haematologica 2000; 85: 47-51
- 392 Sasaki R, Yasuda K, Abe E. et al. Multi-institutional analysis of solitary extramedullary plasmacytoma of the head and neck treated with curative radiotherapy. Int J Radiat Oncol Biol Phys 2012; 82: 626-634
- 393 Knowling MA, Harwood AR, Bergsagel DE. Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. J Clin Oncol 1983; 1: 255-262
- 394 Hamre M, Hedberg J, Buckley J. et al. Langerhans cell histiocytosis: An exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 1997; 28: 92-97
- 395 Laman JD, Leenen PJM, Annels NE. et al. Langerhans-cell histiocytosis „insight into DC biology“. Trends Immunol 2003; 24: 190-196
- 396 Orphanet: Langerhans Zell Histiozytose. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=389
MissingFormLabel
- 397 Kostyra K, Kostkiewicz B. Langerhans cell histiocytosis of the orbit and frontal sinus of the adult woman: A first case report in Poland. Surg Neurol Int 2019; 10: 234
- 398 Mani S, Thomas R, Mathew J. et al. Langerhan’s cell histiocytosis of sphenoid sinus causing vision loss: A case report. J Nepal Med Assoc 2019; 57: 281-284
- 399 Gündüz K, Palamar M, Parmak N. et al. Eosinophilic granuloma of the orbit: Report of two cases. J AAPOS 2007; 11: 506-508
- 400 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthal Plast Reconstr Surg 2016; 32: 28-34
- 401 Prayer D, Grois N, Prosch H. et al. MR imaging presentation of intracranial disease associated with langerhans cell histiocytosis. Am J Neuroradiol 2004; 25: 880-891
- 402 Allen CE, Li L, Peters TL. et al. Cell-Specific Gene Expression in Langerhans Cell Histiocytosis Lesions Reveals a Distinct Profile Compared with Epidermal Langerhans Cells. J Immunol 2010; 184: 4557-4567
- 403 Hyman DM, Diamond EL, Vibat CRT. et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015; 5: 64-71
- 404 Berres ML, Lim KPH, Peters T. et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211: 669-683
- 405 Wladis EJ, Tomaszewski JE, Gausas RE. Langerhans cell histiocytosis of the orbit 10 years after involvement at other sites. Ophthal Plast Reconstr Surg 2008; 24: 142-143
- 406 Herwig MC, Wojno T, Zhang Q. et al. Langerhans Cell Histiocytosis of the Orbit: Five Clinicopathologic Cases and Review of the Literature. Surv Ophthalmol 2013; 58: 330-340
- 407 Windfuhr JP. Primitive neuroectodermal tumor of the head and neck: Incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 2004; 113: 533-543
- 408 Hafezi S, Seethala RR, Stelow EB. et al. Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone. Head Neck Pathol 2011; 5: 8-16
- 409 Specht K, Sung YS, Zhang L. et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged ewing sarcomas: Further evidence toward distinct pathologic entities. Genes Chromosom Cancer 2014; 53: 622-633
- 410 Lin JK, Liang J. Sinonasal Ewing Sarcoma: A Case Report and Literature Review. Perm J 2018; 22
- 411 Allam A, El-Husseiny G, Khafaga Y. et al. Ewing’s sarcoma of the head and neck: A retrospective analysis of 24 cases. Sarcoma 1999; 3: 11-15
- 412 Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184-192
- 413 Broich G, Pagliari A, Ottaviani F. Esthesioneuroblastoma: A general review of the cases published since the discovery of the tumour in 1924. Anticancer Res 1997; 17: 2683-2706
- 414 Roussel LM, Patron V, Maubert E. et al. New landmarks in endonasal surgery: from nasal bone to anterior cribriform plate including branches of anterior ethmoidal artery and nerve and terminal nerve. Int Forum Allergy Rhinol 2020; 10: 395-404
- 415 Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 2002; 15: 264-278
- 416 Thompson LDR. Olfactory Neuroblastoma. Head Neck Pathol 2009; 3: 252-259
- 417 Kadish S, Goodman M, Wang CC. Olfactory neuroblastoma—A clinical analysis of 17 cases. Cancer 1976; 37: 1571-1576
- 418 Morita A, Ebersold MJ, Olsen KD. et al. Esthesioneuroblastoma: Prognosis and management. Neurosurgery 1993; 32: 706-715
- 419 Edge SB, Compton CC. The american joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010; 17: 1471-1474
- 420 Norton A. Tumors of the Upper Respiratory Tract and Ear. In: Hyams V, Batsakis J, Michaels L Hrsg. Journal of Clinical Pathology 1989; 335-335
- 421 Mao L, Xia YP, Zhou YN. et al. Activation of sonic hedgehog signaling pathway in olfactory neuroblastoma. Oncology 2009; 77: 231-243
- 422 Ketcham AS, Wilkins RH, Van Buren JM. et al. A combined intracranial facial approach to the paranasal sinuses. Am J Surg 1963; 106: 698-703
- 423 Hanna E, DeMonte F, Ibrahim S. et al. Endoscopic resection of sinonasal cancers with and without craniotomy: Oncologic results. Arch Otolaryngol - Head Neck Surg 2009; 135: 1219-1224
- 424 Su SY, Bell D, Ferrarotto R. et al. Outcomes for olfactory neuroblastoma treated with induction chemotherapy. Head Neck 2017; 39: 1671-1679
- 425 Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: A meta-analysis and review. Lancet Oncol 2001; 2: 683-690
- 426 Ow TJ, Hanna EY, Roberts DB. et al. Optimization of long-term outcomes for patients with esthesioneuroblastoma. Head Neck 2014; 36: 524-530
- 427
Almutuawa DM,
Strohl MP,
Gruss C.
et al. Outcomes of sinonasal mucosal melanomas with endoscopic and open resection:
a
retrospective cohort study. J Neurooncol. 2020
MissingFormLabel
- 428 Thompson LDR, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: A clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol 2003; 27: 594-611
- 429 Moreno MA, Roberts DB, Kupferman ME. et al. Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson cancer center. Cancer 2010; 116: 2215-2223
- 430 Patel SG, Prasad ML, Escrig M. et al. Primary mucosal malignant melanoma of the head and neck. Head Neck 2002; 24: 247-257
- 431 Zebary A, Jangard M, Omholt K. et al. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: A study of 56 cases. Br J Cancer 2013; 109: 559-564
- 432 Van Essen TH, Van Pelt S, Versluis M. et al. Prognostic parameters in uveal melanoma and their association with bap1 expression. Br J Ophthalmol 2014; 98: 1738-1743
- 433 Dauer EH, Lewis JE, Rohlinger AL. et al. Sinonasal melanoma: A clinicopathologic review of 61 cases. Otolaryngol - Head Neck Surg 2008; 138: 347-352
- 434 Bachar G, Kwok SL, O’Sullivan B. et al. Mucosal melanomas of the head and neck: The Princess Margaret Hospital experience. Head Neck 2008; 30: 1325-1331
- 435
Lund VJ,
Chisholm EJ,
Howard DJ.
et al. Sinonasal malignant melanoma: An analysis of 115 cases assessing outcomes of
surgery, postoperative radiotherapy and endoscopic resection. Rhinology 2012; 50:
203-210
MissingFormLabel
- 436 Thierauf J, Glück AM, Plinkert P. et al. Mucosal melanoma of the cranio-facial region: Surgical challenges and therapeutic options. Auris Nasus Larynx 2019; 46: 252-259
- 437 Gore MR, Zanation AM. Survival in sinonasal melanoma: A meta-analysis. J Neurol Surgery, Part B Skull Base 2012; 73: 157-162
- 438 Ganly I, Patel SG, Singh B. et al. Craniofacial resection for malignant melanoma of the skull base: Report of an International Collaborative Study. Arch Otolaryngol - Head Neck Surg 2006; 132: 73-78
- 439 Jarrom D, Paleri V, Kerawala C. et al. Mucosal melanoma of the upper airways tract mucosal melanoma: A systematic review with meta-analyses of treatment. Head Neck 2017; 39: 819-825
- 440 Li J, Kan H, Zhao L. et al. Immune checkpoint inhibitors in advanced or metastatic mucosal melanoma: a systematic review. Ther Adv Med Oncol 2020; 12
- 441 Misra P, Husain Q, Svider PF. et al. Management of sinonasal teratocarcinosarcoma: A systematic review. Am J Otolaryngol - Head Neck Med Surg 2014; 35: 5-11
- 442 Chao KK, Eng TY, Barnes J. et al. Sinonasal Teratocarcinosarcoma. Am J Clin Oncol Cancer Clin Trials 2004; 27: 29-32
- 443 Fukuoka K, Hirokawa M, Shimizu M. et al. Teratocarcinosarcoma of the nasal cavity. Report of a case showing favorable prognosis. Apmis 2000; 108: 553-557
- 444 Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses: A clinicopathologic study of 20 cases. Cancer 1984; 53: 2140-2154
- 445 Wei S, Carroll W, Lazenby A. et al. Sinonasal teratocarcinosarcoma: report of a case with review of literature and treatment outcome. Ann Diagn Pathol 2008; 12: 415-425
- 446 Vranic S, Caughron SK, Djuricic S. et al. Hamartomas, teratomas and teratocarcinosarcomas of the head and neck: Report of 3 new cases with clinico-pathologic correlation, cytogenetic analysis, and review of the literature. BMC Ear, Nose Throat Disord 2008; 8
- 447 Lim CCT, Thiagarajan A, Sim CS. et al. Craniospinal dissemination in teratocarcinosarcoma: Case report. J Neurosurg 2008; 109: 321-324
- 448 Terasaka S, Medary MB, Whiting DM. et al. Prolonged survival in a patient with sinonasal teratocarcinosarcoma with cranial extension. Case report. J Neurosurg 1998; 88: 753-756
- 449 Dicke TE, Gates GA. Malignant Teratoma of the Paranasal Sinuses: Report of a Case. Arch Otolaryngol 1970; 91: 391-394
Publication History
Article published online:
30 April 2021
© 2021. The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution-NonDerivative-NonCommercial-License, permitting copying and reproduction so long as the original work is given appropriate credit. Contents may not be used for commercial purposes, or adapted, remixed, transformed or built upon. (https://creativecommons.org/licenses/by-nc-nd/4.0/).
Georg Thieme Verlag KG
Rüdigerstraße 14, 70469 Stuttgart, Germany
-
Literaturverzeichnis
- 1 Bundesministerium für Gesundheit Seltene Erkrankungen - Bundesgesundheitsministerium.
2019 Im Internet: https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
MissingFormLabel
- 2 Anzahl I. Prävalenzen en und Inzidenzen seltener Krankheiten: Bibliografische Angaben. 2015; 1-52 . Im Internet: https://www.orpha.net/orphacom/cahiers/docs/DE/Pravalenzen_seltener_Krankheiten_Alphabetische_Liste.pdf
- 3 Pedan H, Janosova V, Hajtman A, Calkovsky V. Non-Reflex Defense Mechanisms of Upper Airway Mucosa: Possible Clinical Application. Physiol Res 2020; 69 (Suppl. 01) S55-S67 PMID: 32228012
- 4 Sommer F, Scheithauer M, Kröger R. et al. Niesen als mechanische Abwehr – Numerische Simulation mit Analyse der durchströmten Nasenbereiche. Laryngorhinootologie 2014; 93: 746-750
- 5 Sommer F, Hoffmann TK, Mlynski G. et al. Dreidimensionale Analyse nasaler PhysiologieThree-dimensional analysis of nasal physiology. HNO 2017; 66: 280-289
- 6 Lindemann J, Reichert M, Kröger R. et al. Numerical simulation of humidification and heating during inspiration in nose models with three different located septal perforations. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1795-1800
- 7 Sommer F, Kröger R, Lindemann J. Numerical simulation of humidification and heating during inspiration within an adult nose. Rhinology 2012; 50: 157-164
- 8 Lindemann J, Rettinger G, Kröger R. et al. Numerical simulation of airflow patterns in nose models with differently localized septal perforations. Laryngoscope 2013; 123: 2085-2089
- 9 Götte K, Hörmann K. Sinonasal malignancy: What’s new?. Orl 2004; 66: 85-97
- 10 Barnes L, Eveson JW, Reichart P. et al. World health organization classification of tumours: Pathology and genetics of head and neck tumours. Lyon: International Agency for Research on Cancer; 2005
- 11 Slinger CA, McGarry GW. Nose and sinus tumours: Red flags and referral. Br J Gen Pract 2018; 68: 247-248
- 12 Le QT, Fu KK, Kaplan M. et al. Treatment of maxillary sinus carcinoma: A comparison of the 1997 and 1977 American Joint Committee on Cancer staging systems. Cancer 1999; 86: 1700-1711
- 13 Tiwari R, Hardillo JA, Mehta D. et al. Squamous cell carcinoma of maxillary sinus. Head Neck 2000; 22: 164-169
- 14 Tiwari R, Hardillo JA, Tobi H. et al. Carcinoma of the ethmoid: Results of treatment with conventional surgery and post-operative radiotherapy. Eur J Surg Oncol 1999; 25: 401-405
- 15 Laudien M. Ausgewählte seltene rhinologische Krankheitsbilder Pathogenese – Klinik – Therapie. Laryngorhinootologie 2015; 94: S272-S287
- 16 Hauser LJ, Jensen EL, Mirsky DM. et al. Pediatric anosmia: A case series. Int J Pediatr Otorhinolaryngol 2018; 110: 135-139
- 17 Hummel T, Whitcroft KL, Andrews P. et al. Position paper on olfactory dysfunction. Rhinol J 2017; 0: 1-30
- 18 Orphanet: Anosmie, isolierte kongenitale. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=11807&Disease_Disease_Search_diseaseGroup=anosmie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Anosmie--isolierte-kongenitale&title=Anosmie
isolierte kongenit
MissingFormLabel
- 19 Mahmood AN, Abulaban O, Janjua A. Bilateral olfactory aplasia: Uncommon cause of congenital anosmia. Clin Case Reports 2019; 7: 1456-1457
- 20 Orphanet: Kallmann Syndrom. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=478
MissingFormLabel
- 21 Johnson VP, McMillin JM, Aceto T. et al. A newly recognized neuroectodermal syndrome of familial alopecia, anosmia, deafness, and hypogonadism. Am J Med Genet 1983; 15: 497-506
- 22 Gebril O, Abdelraouf E, Shafie M. et al. Johnson-McMillin microtia syndrome: New additional family. J Fam Med Prim Care 2014; 3: 275
- 23 Orphanet: Arrhinie, isolierte. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert=1134
MissingFormLabel
- 24 Thiele H, Musil A, Nagel F. et al. Familial arhinia, choanal atresia, and microphthalmia. Am J Med Genet 1996; 63: 310-313
- 25 Abukhalaf SA, Zalloum JS, Al Hammouri A. et al. Congenital arrhinia: A case report and literature review. Int J Pediatr Otorhinolaryngol 2020; 135: 110083
- 26 Jung JW, Ha DH, Kim BY. et al. Nasal Reconstruction Using a Customized Three-Dimensional–Printed Stent for Congenital Arhinia: Three-Year Follow-up. Laryngoscope 2019; 129: 582-585
- 27 Núñez-Villaveiran T, Frohner BB, Urcelay PR. et al. Bifid nose – A mild degree of frontonasal dysplasia. A case report. Int J Pediatr Otorhinolaryngol 2013; 77: 1374-1377
- 28 Orphanet: Bifid nose. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=2457&Disease_Disease_Search_diseaseGroup=bifid-nose&Disease_Disease_Search_diseaseType=Pat&Disease(s)/groupofdiseases=Bifid-nose&title=Bifid
nose&search=Disease_Search_Simple
MissingFormLabel
- 29 Sedano HO, Michael Cohen M, Jirasek J. et al. Frontonasal dysplasia. J Pediatr 1970; 76: 906-913
- 30 Genç E, Derbent M, Ergin NT. A mild case of frontonasal dysplasia: The rhinologic perspective. Int J Pediatr Otorhinolaryngol 2002; 65: 75-83
- 31 Cohen MM. Perspectives on craniosynostosis: Sutural biology, some well-known syndromes, and some unusual syndromes. J Craniofac Surg 2009; 20: 646-651
- 32 Orphanet: Kraniorhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=de&Expert=157832
MissingFormLabel
- 33 Rüegg EM, Bartoli A, Rilliet B. et al. Management of median and paramedian craniofacial clefts. J Plast Reconstr Aesthetic Surg 2019; 72: 676-684
- 34 Orphanet: Paramedian nasal cleft. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=17067&Disease_Disease_Search_diseaseGroup=paramediannasal-cleft&Disease_Disease_Search_diseasType=Pat&Disease(s)/groupofdiseases=Paramedian-nasal-cleft&title=Paramedian%20nasal%20cleft
MissingFormLabel
- 35 Williams A, Pizzuto M, Brodsky L. et al. Supernumerary nostril: A rare congenital deformity. Int J Pediatr Otorhinolaryngol 1998; 44: 161-167
- 36 Orphanet: Polyrhinie. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17033&Disease_Disease_Search_diseaseGroup=Polyrhinie&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Polyrhinie&title=Polyrhinie&search=Disease_Search_Simple
MissingFormLabel
- 37 Orphanet: Nasenlöcher, überzählige. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=17034&Disease_Disease_Search_diseaseGroup=Nasenloch-&Disease_Disease_Search_diseaseType=Pat&Krankheite(n)/Krankheitsgruppe=Nasenlocher--uberzahlige&title=Nasenl%F6cher,%FCberz%E4hlige
MissingFormLabel
- 38 Gulasi S, Turhan AH, Celik Y. et al. Accessory nostril: A rare congenital nasal anomaly. J Craniofac Surg 2015; 26: e602-e603
- 39 Galiè M, Clauser LC, Tieghi R. et al. The arrhinias: Proboscis lateralis literature review and surgical update. J Cranio-Maxillofacial Surg 2019; 47: 1410-1413
- 40 Harada T, Muraoka M. Proboscis lateralis: A rare bilateral case [7]. Ann Plast Surg 2001; 47: 350-351
- 41 Wolfe SA, Tessier P, Ciminello FS. The Arrhinias. Scand J Plast Reconstr Surg Hand Surg 2009; 43: 177-196
- 42 Rosen Z, Gitlin G. Bilateral Nasal Proboscis: Associated with Unilateral Anophthalmia, Unilateral Diffuse Pigmentation of the Conjunctiva, and Anomalies of the Skull and Brain. AMA Arch Otolaryngol 1959; 70: 545-550
- 43 El-fattah AMA, Naguib A, El-Sisi H. et al. Midline nasofrontal dermoids in children: A review of 29 cases managed at Mansoura University Hospitals. Int J Pediatr Otorhinolaryngol 2016; 83: 88-92
- 44 Orphanet: Nasal dorsum fistula. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=141219
MissingFormLabel
- 45 Jurkov M, Olze H, Klauschen F. et al. IgG4-related orbitopathy as an important differential diagnosis of advanced silent sinus syndrome. HNO 2020; 68: 65-68
- 46 MONTGOMERY WW. Mucocele of the Maxillary Sinus Causing Enophthalmos. Eye Ear Nose Throat Mon 1964; 43: 41-44
- 47 Soparkar CNS, Patrinely JR, Cuaycong MJ. et al. The Silent Sinus Syndrome: A Cause of Spontaneous Enophthalmos. Ophthalmology 1994; 101: 772-778
- 48 Naik RM, Khemani S, Saleh HA. Frontal silent sinus syndrome. Otolaryngol - Head Neck Surg (United States) 2013; 148: 354-355
- 49 McArdle B, Perry C. Ethmoid silent sinus syndrome causing inward displacement of the orbit: Case report. J Laryngol Otol 2010; 124: 206-208
- 50 Wise SK, Wojno TH, DelGaudio JM. Silent sinus syndrome: Lack of orbital findings in early presentation. Am J Rhinol 2007; 21: 489-494
- 51 Brandt MG, Wright ED. The silent sinus syndrome is a form of chronic maxillary atelectasis: A systematic review of all reported cases. Am J Rhinol 2008; 22: 68-73
- 52 Rose GE, Lund VJ. Clinical features and treatment of late enophthalmos after orbital decompression: A condition suggesting cause for idiopathic „imploding antrum“ (silent sinus) syndrome. Ophthalmology 2003; 110: 819-826
- 53 Numa WA, Desai U, Gold DR. et al. Silent sinus syndrome: A case presentation and comprehensive review of all 84 reported cases. Ann Otol Rhinol Laryngol 2005; 114: 688-694
- 54 Davidson JK, Soparkar CNS, Williams JB. et al. Negative sinus pressure and normal predisease imaging in silent sinus syndrome. Arch Ophthalmol 1999; 117: 1653-1654
- 55 Annino DJ, Goguen LA. Silent sinus syndrome. Curr Opin Otolaryngol Head Neck Surg 2008; 16: 22-25
- 56 Hildenbrand T, Klein SB, Schramek N. et al. Rare diseases of the maxillary sinus. HNO 2020; 68: 581-589
- 57 Facon F, Eloy P, Brasseur P. et al. The silent sinus syndrome. Eur Arch Oto-Rhino-Laryngology 2006; 263: 567-571
- 58 Hourany R, Aygun N, Della Santina CC. et al. Silent sinus syndrome: An acquired condition. Am J Neuroradiol 2005; 26: 2390-2392
- 59 Lee DS, Murr AH, Kersten RC. et al. Silent sinus syndrome without opacification of ipsilateral maxillary sinus. Laryngoscope 2018; 128: 2004-2007
- 60 Illner A, Davidson HC, Harnsberger HR. et al. The silent sinus syndrome: Clinical and radiografic findings. Am J Roentgenol 2002; 178: 503-506
- 61 Rose GE, Sandy C, Hallberg L. et al. Clinical and radiologic characteristics of the imploding antrum, or „silent sinus,“ syndrome. Ophthalmology 2003; 110: 811-818
- 62 Weber RK. Aktueller Stand der endonasalen Nasennebenhöhlenchirurgie
[Comprehensive review on endonasal endoscopic sinus
surgery]. Laryngorhinootologie. 2015; 94 Suppl 1; S64–S142.
doi:110.1055/s-0035-1545353. Epub 2015 Apr 10. PMID: 25860497
MissingFormLabel
- 63 Sogg A. Endoscopic sinus surgery. 4th Editio Thieme Verlag; 1993
- 64 Sommer F, Hoffmann T, Lindemann J. et al. Radicality of maxillary sinus surgery and size of the maxillary sinus ostium. HNO 2020; 68: 573-580
- 65 Thomas RD, Graham SM, Carter KD. et al. Management of the orbital floor in silent sinus syndrome. Am J Rhinol 2003; 17: 97-100
- 66 Babar-Craig H, Kayhanian H, de Silva DJ. et al. Spontaneous silent sinus syndrome (Imploding antrum syndrome): Case series of 16 patients. Rhinology 2011; 49: 315-317
- 67 Sivasubramaniam R, Sacks R, Thornton M. Silent sinus syndrome: Dynamic changes in the position of the orbital floor after restoration of normal sinus pressure. J Laryngol Otol 2011; 125: 1239-1243
- 68 Adams WM, Jones RI, Chavda SI. et al. Pneumosinus dilatans: a discussion of four cases and the possible aetiology. Rhinology 1998; 36: 40-402
- 69 Urken ML, Som PM, Lawson W. et al. Abnormally large frontal sinus. II. Nomenclature, pathology, and symptoms. Laryngoscope 1987; 97: 606-611
- 70 Kim WJ, Kim MM. Binasal hemianopia caused by pneumosinus dilatans of the sphenoid sinuses. Indian J Ophthalmol 2019; 67: 1772-1775
- 71 Skolnick CA, Mafee MF, Goodwin JA. Pneumosinus Dilatans of the Sphenoid Sinus Presenting With Visual Loss. J Neuro-ophthalmology 2001; 20: 259-263
- 72 Alatar AA, AlSuliman YA, Alrajhi MS. et al. Maxillary Pneumosinus Dilatans Presenting With Proptosis: A Case Report and Review of the Literature. Clin Med Insights Ear, Nose Throat 2019; 12: 117955061882514
- 73 Sweatman J, Beltechi R. Pneumosinus Dilatans: An exploration into the association between Arachnoid Cyst, Meningioma and the pathogenesis of Pneumosinus Dilatans. Clin Neurol Neurosurg 2019; 185
- 74 Ricci JA. Pneumosinus Dilatans: Over 100 Years Without an Etiology. J Oral Maxillofac Surg 2017; 75: 1519-1526
- 75 Martin AJ, Jarosz JM, Thomas NWM. The strange association of pneumosinus dilatans and arachnoid cyst: Case report and review of the literature. Acta Neurochir (Wien) 2001; 143: 197-201
- 76 Gibbons BA, Miele WR, Florman JE, Heilman CB, Horgan MA
Pneumosinus dilitans and meningioma: a case series and review of
the literature. Neurosurg Focus 2011; 50(5): E13. doi:10.3171/2011.
3.FOCUS1113. PMID: 21529169
MissingFormLabel
- 77 Desai NS, Saboo SS, Khandelwal A. et al. Pneumosinus dilatans: Is it more than an aesthetic concern?. J Craniofac Surg 2014; 25: 418-421
- 78 Draf W, Constantinidis J, Weber R. et al. Pneumosinus Dilatans Frontalis, Atiologie, Symptomatik Und Operationstechnik. Laryngorhinootologie 1996; 75: 660-664
- 79 Cho HS, Hong SJ, Chae HK. et al. Maxillary Sinus Pneumocele Presenting as Aesthetic Deformity: A Case Report With Literature Review. Ear, Nose Throat J 2020; 99: 397-401
- 80 Song M, Ahn SM, Reh DR. et al. Ethmoid pneumocele presenting with exophthalmos 15 years after endoscopic sinus surgery. Allergy Rhinol 2015; 6: 129-132
- 81 Braverman I. Pneumocele of the maxillary sinus with orbital and
trigeminal nerve involvement: case report and review of the
literature. J Otolaryngol Head Neck Surg 2009; 38(2): E35–E38.
PMID: 19442351
MissingFormLabel
- 82 Sasaki T, Yamoto T, Fujita K. et al. A Case of Orbital Emphysema Associated with Frontal Sinus Pneumocele. J Neurol Surg Reports 2013; 74: 054-056
- 83 Bachor E, Weber R, Kahle G. et al. Temporary unilateral amaurosis with pneumosinus dilatans of the sphenoid sinus. Skull Base Surg 1994; 4: 169-175
- 84 Bell AF, Ivan DJ, Munson RA. Barosinus pneumocele: Transient visual loss due to sphenoid sinus pneumocele in a U.S. Air Force pilot. Aviat Sp Environ Med 1995; 66: 276-279
- 85 Choi SJ, Seo ST, Rha KS. et al. Sinonasal organized Hematoma: Clinical features of seventeen cases and a systematic review. Laryngoscope 2015; 125: 2027-2033
- 86 Kim JS, Oh JS, Kwon SH. The increasing incidence of paranasal organizing hematoma: a 20-year experience of 23 cases at a single center. Rhinol J 2016; 54: 176-182
- 87 Pang W, Hu L, Wang H. et al. Organized Hematoma: An Analysis of 84 Cases with Emphasis on Difficult Prediction and Favorable Management. Otolaryngol – Head Neck Surg (United States) 2016; 154: 626-633
- 88 Song HM, Jang YJ, Chung YS. et al. Organizing hematoma of the maxillary sinus. Otolaryngol - Head Neck Surg 2007; 136: 616-620
- 89 Young D. Surgical treatment of male infertility. J Reprod Fertil 1970; 23: 541-542
- 90 Hendry WF, Knight RK, Whitfield HN. et al. Obstructive Azoospermia: Respiratory Function Tests, Electron Microscopy and the Results of Surgery. Br J Urol 1978; 50: 598-604
- 91 Arya AK, Beer HL, Benton J. et al. Does Young’s syndrome exist?. J Laryngol Otol 2009; 123: 477-481
- 92 Hendry WF, Levison DA, Parkinson MC. et al. Testicular obstruction: Clinicopathological studies. Ann R Coll Surg Engl 1990; 72: 396-407
- 93 Shiraishi K, Ono N, Eguchi S. et al. Young’s syndrome associated with situs inversus totalis. Arch Androl 2004; 50: 169-172
- 94 Hasegawa A, Fujita M, Ohe M. et al. A Rare Case of Young’s Syndrome in Japan. Intern Med 1994; 33: 649-653
- 95 Armengot M, Juan G, Carda C. et al. Young’s syndrome: A further cause of chronic rhinosinusitis. Rhinology 1996; 34: 35-37
- 96 Greenstone MA, Rutman A, Hendry WF. et al. Ciliary function in Young’s syndrome. Thorax 1988; 43: 153-154
- 97 Stern BM, Sharma G. Ciliary Dysfunction (Kartagener Syndrome, Primary Ciliary Dyskinesia). StatPearls Publishing; 2019
- 98 Samia H, Khadija B, Agnes H. et al. Long-term outcome of tunisian children with primary ciliary dyskinesia confirmed by transmission electron microscopy. Afr Health Sci 2016; 16: 954-961
- 99 Schofield LM, Duff A, Brennan C. Airway Clearance Techniques for Primary Ciliary Dyskinesia; is the Cystic Fibrosis literature portable?. Paediatr Respir Rev 2018; 25: 73-77
- 100 Alanin MC, Aanaes K, Høiby N. et al. Sinus surgery can improve quality of life, lung infections, and lung function in patients with primary ciliary dyskinesia. Int Forum Allergy Rhinol 2017; 7: 240-247
- 101 Nyilas S, Schlegtendal A, Singer F, Goutaki M, Kuehni CE, Casaulta C,
Latzin P, Koerner-Rettberg C. Alternative inert gas washout outcomes
in patients with primary ciliary dyskinesia. Eur Respir J 2017; 49(1):
1600466. doi:10.1183/13993003.00466-2016. PMID: 28122863
MissingFormLabel
- 102 Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, Dell S,
Eber E, Escudier E, Hirst RA, Hogg C, Jorissen M, Latzin P, Legendre M,
Leigh MW, Midulla F, Nielsen KG, Omran H, Papon JE, Pohunek P,
Redfern B, Rigau D, Rindlisbacher B, Santamaria F, Shoemark A,
Snijders D, Tonia T, Titieni A, Walker WT, Werner C, Bush A, Kuehni
CE. European Respiratory Society guidelines for the diagnosis of
primary ciliary dyskinesia. Eur Respir J 2017; 49(1); 1601090.
doi:10.1183/13993003.00466-2016. PMID: PMC6054534
MissingFormLabel
- 103 Strippoli MPF, Frischer T, Barbato A. et al. Management of primary ciliary dyskinesia in European children: Recommendations and clinical practice. Eur Respir J 2012; 39: 1482-1491
- 104 Barbato A, Frischer T, Kuehni CE. et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. In: European Respiratory Journal. Eur Respir J 2009; 1264-1276
- 105 Lucas JS, Burgess A, Mitchison HM. et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child 2014; 99: 850-856
- 106 El-Naggar AK, Chan JKC, Grandis JR et al. WHO Classification of Head
and Neck Tumours. 4th Edition Lyon: International Agency for
Research on Cancer; 2017
MissingFormLabel
- 107 Lombardi D, Tomenzoli D, Buttà L. et al. Limitations and complications of endoscopic surgery for treatment for sinonasal inverted papilloma: A reassessment after 212 cases. Head Neck 2011; 33: 1154-1161
- 108 Stange T, Schultz-Coulon HJ. Zum Chirurgischen Behandlungskonzept bei Invertierten Papillomen der Nase und Nasennebenhöhlen. HNO 2008; 56: 614-622
- 109 Christensen WN, Smith RRL. Schneiderian papillomas. A clinicopathologic study of 67 cases. Hum Pathol 1986; 17: 393-400
- 110 Stammberger H. New aspects in the genesis of inverted papillomas. Laryngol Rhinol Otol (Stuttg) 1983; 62: 249-24955
- 111 Leoncini G, Zanetti L. The papillomas of the sinonasal tract. A comprehensive review. Pathologica 2017; 109: 31-34
- 112 Sarkar FH, Visscher DW, Kintanar EB. et al. Sinonasal Schneiderian papillomas: human papillomavirus typing by polymerase chain reaction. Mod Pathol 1992; 5: 329-332
- 113 Barnes L. Schneiderian papillomas and nonsalivary glandular neoplasms of the head and neck. Mod Pathol 2002; 15: 279-297
- 114 Cunningham MJ, Brantley S, Barnes L. et al. Oncocytic Schneiderian papilloma in a young adult: A rare diagnosis. Otolaryngol Neck Surg 1987; 97: 47-51
- 115 Barnes L, Bedetti C. Oncocytic schneiderian papilloma: A reappraisal of cylindrical cell papilloma of the sinonasal tract. Hum Pathol 1984; 15: 344-351
- 116 Michaels L, Young M. Histogenesis of papillomas of the nose and paranasal sinuses. Arch Pathol Lab Med 1995; 119: 821-826
- 117 Lawson W, Patel ZM. The evolution of management for inverted papilloma: An analysis of 200 cases. Otolaryngol – Head Neck Surg 2009; 140: 330-335
- 118 Lund VJ, Stammberger H, Nicolai P. et al. European position paper on endoscopic management of tumours of the nose, paranasal sinuses and skull base. Rhinol Suppl 2010; 1-143
- 119 Maitra A, Baskin LB, Lee EL. Malignancies arising in oncocytic schneiderian papillomas: A report of 2 cases and review of the literature. Arch Pathol Lab Med 2001; 125: 1365-1367
- 120 Anari S, Carrie S. Sinonasal inverted papilloma: Narrative review. J Laryngol Otol 2010; 124: 705-715
- 121 Attlmayr B, Derbyshire SG, Kasbekar AV et al. Management of
inverted papilloma: Review. In: Journal of Laryngology and Otology.
Cambridge University Press; 2017: 284–289
MissingFormLabel
- 122 Heathcote KJ, Nair SB. The impact of modern techniques on the recurrence rate of inverted papilloma treated by endonasal surgery. Rhinology 2009; 47: 339-344
- 123 Diamantopoulos II, Jones NS, Lowe J. All nasal polyps need histological examination: An audit-based appraisal of clinical practice. J Laryngol Otol 2000; 114: 755-759
- 124 Lloyd GAS, Lloyd GAS. Epithelial Tumours. In: Diagnostic Imaging of the Nose and Paranasal Sinuses 1988; 95-110
- 125 Hildenbrand T, Weber R, Mertens J. et al. Surgery of Inverted Papilloma of the Maxillary Sinus via Translacrimal Approach – Long-Term Outcome and Literature Review. J Clin Med 2019; 8: 1873
- 126 Giotakis E, Knipping S, Kühnel T. et al. Unilateral diseases of the maxillary sinus. HNO 2020; 68: 623-636
- 127 Vrabec DP. The inverted schneiderian papilloma: A 25-year study. Laryngoscope 1994; 104: 582-605
- 128 Bielamowicz S, Calcaterra TC, Watson D. Inverting papilloma of the head and neck: The UCLA update. Otolaryngol – Head Neck Surg 1993; 109: 71-76
- 129 Kraft M, Simmen D, Kaufmann T. et al. Long-term results of endonasal sinus surgery in sinonasal papillomas. Laryngoscope 2003; 113: 1541-1547
- 130 Tanna N, Edwards JD, Aghdam H. et al. Transnasal endoscopic medial maxillectomy as the initial oncologic approach to sinonasal neoplasms: The anatomic basis. Arch Otolaryngol – Head Neck Surg 2007; 133: 1139-1142
- 131 Busquets JM, Hwang PH. Endoscopic resection of sinonasal inverted papilloma: A meta-analysis. Otolaryngol – Head Neck Surg 2006; 134: 476-482
- 132 Veeresh M, Sudhakara M, Girish G. et al. Leiomyoma: A rare tumor in the head and neck and oral cavity: Report of 3 cases with review. J Oral Maxillofac Pathol 2013; 17: 281-287
- 133 Agaimy A, Michal M, Thompson LDR. et al. Angioleiomyoma of the Sinonasal Tract: Analysis of 16 Cases and Review of the Literature. Head Neck Pathol 2015; 9: 463-473
- 134 Heyman JNS, Jones LM, Hilton JM. et al. Angioleiomyoma of the inferior turbinate: A rare cause of isolated facial pain. Ann R Coll Surg Engl 2020; 102: E20-E22
- 135 Lau YW, Tharumalingam V, Tan TY. et al. Sinonasal angioleiomyoma. Med J Malaysia 2016; 71: 154-155
- 136 Mathieu T, Verbruggen A, Goovaerts G. et al. Vascular leiomyoma of the nasal cavity: case report and literature review. Eur Arch Oto-Rhino-Laryngology 2017; 274: 579-583
- 137 Poncet A, Dor L. Botryomycose humaine. Rev Chir 1897; 18: 996-1003
- 138 Hartzell M. Granuloma pyogenicum (botryomycosis of French authors). J Cutan Dis 1904; 22: 520-523
- 139 Mills SE, Cooper PH, Fechner RE. Lobular capillary hemangioma: The underlying lesion of pyogenic granuloma. A study of 73 cases from the oral and nasal mucous membranes. Am J Surg Pathol 1980; 4: 471-479
- 140 Fu Y-S, Perzin KH. Nonepithelial tumors of the nasal cavity, paranasal sinuses, and nasopharynx. A clinicopathologic study VI. Fibrous tissue tumors (fibroma, fibromatosis, fibrosarcoma). Cancer 1976; 37: 2912-2928
- 141 Thompson LDR, Fanburg-Smith JC. Update on Select Benign Mesenchymal and Meningothelial Sinonasal Tract Lesions. Head Neck Pathol 2016; 10: 95-108
- 142 Sammut SJ, Tomson N, Corrie P. Pyogenic granuloma as a cutaneous adverse effect of vemurafenib. N Engl J Med 2014; 371: 1265-1267
- 143 Smith SC, Patel RM, Lucas DR. et al. Sinonasal Lobular Capillary Hemangioma: A Clinicopathologic Study of 34 Cases Characterizing Potential for Local Recurrence. Head Neck Pathol 2013; 7: 129-134
- 144 Sunaryo PL, Svider PF, Husain Q. et al. Schwannomas of the sinonasal tract and anterior skull base: A systematic review of 94 cases. Am J Rhinol Allergy 2014; 28: 39-49
- 145 Shugar JMA, Son PM, Biller HF. et al. Peripheral nerve sheath tumors of the paranasal sinuses. Head Neck Surg 1981; 4: 72-76
- 146 Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg 1994; 81: 362-373
- 147 Ansari I, Ansari A, Graison AA. et al. Head and Neck Schwannomas: A Surgical Challenge – A Series of 5 Cases. Case Rep Otolaryngol 2018; 2018: 1-10
- 148 Azani AB, Bishop JA, Thompson LDR. Sinonasal Tract Neurofibroma: A Clinicopathologic Series of 12 Cases with a Review of the Literature. Head Neck Pathol 2015; 9: 323-333
- 149 Annino DJ, Domanowski GF, Vaughan CW. A Rare Cause of Nasal Obstruction: A Solitary Neurofibroma. Otolaryngol Neck Surg 1991; 104: 484-488
- 150 Perzin KH, Panyu H, Wechter S. Nonepithelial tumors of the nasal cavity, paranasal sinuses and nasopharynx. A clinicopathologic study. XII: Schwann cell tumors (neurilemoma, neurofibroma, malignant schwannoma). Cancer 1982; 50: 2193-2202
- 151 Friedman CD, Costantino PD, Teitelbaum B. et al. Primary extracranial meningiomas of the head and neck. Laryngoscope 1990; 100: 41-48
- 152 Thompson LDR, Gyure KA. Extracranial sinonasal tract meningiomas: A clinicopathologic study of 30 cases with a review of the literature. Am J Surg Pathol 2000; 24: 640-650
- 153 Ho K-L. Primary meningioma of the nasal cavity and paranasal sinuses. Cancer 1980; 46: 1442-1447
- 154 Perry A, Scheithauer BW, Stafford SL. et al. „Malignancy“ in meningiomas: A clinicopathologic study of 116 patients, with grading implications. Cancer 1999; 85: 2046-2056
- 155 Perry A, Stafford SL, Scheithauer BW. et al. Meningioma grading: An analysis of histologic parameters. Am J Surg Pathol 1997; 21: 1455-1465
- 156 Rushing EJ, Bouffard JP, McCall S. et al. Primary extracranial meningiomas: An analysis of 146 cases. Head Neck Pathol 2009; 3: 116-130
- 157 Mnejja M, Hammami B, Bougacha L. et al. Primary sinonasal meningioma. Eur Ann Otorhinolaryngol Head Neck Dis 2012; 129: 47-50
- 158
Wolf A,
Safran B,
Pock J.
et al. Surgical treatment of paranasal sinus osteomas: A single center experience
of 58
cases. Laryngoscope. 2019
MissingFormLabel
- 159 Atallah N, Jay MM. Osteomas of the paranasal sinuses. J Laryngol Otol 1981; 95: 291-304
- 160 Furlaneto EC, Rocha JRM, Heitz C. Osteoma of the zygomatic arch - Report of a case. Int J Oral Maxillofac Surg 2004; 33: 310-311
- 161 Summers LE, Mascott CR, Tompkins JR. et al. Frontal sinus osteoma associated with cerebral abscess formation: A case report. Surg Neurol 2001; 55: 235-239
- 162 Eller R, Sillers M. Common Fibro-osseous Lesions of the Paranasal Sinuses. Otolaryngol Clin North Am 2006; 39: 585-600
- 163 Schick B, Steigerwald C, El Tahan AER. et al. The role of endonasal surgery in the management of frontoethmoidal osteomas. Rhinology 2001; 39: 66-70
- 164 Schick B, Dlugaiczyk J. Benign tumors of the nasal cavity and paranasal sinuses. In: Rhinology and Facial Plastic Surgery 2009; 377-385
- 165 Buyuklu F, Akdogan MV, Ozer C. et al. Growth characteristics and clinical manifestations of the paranasal sinus osteomas. Otolaryngol - Head Neck Surg 2011; 145: 319-323
- 166 Golant A, Zeichner JA. Gardner Syndrome. StatPearls Publishing; 2014
- 167 Pinto RS, Simons A, Verma R. et al. Gardener-associated fibroma: An unusual cause of upper airway obstruction. BMJ Case Rep 2018; 2018
- 168 Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations
in hepatoblastoma. Pediatr Blood Cancer 2018; 65(4); doi:10.1002/
pbc.26892. Epub 2017 Dec 18. PMID: 29251405
MissingFormLabel
- 169 Kiessling P, Dowling E, Huang Y, Ho ML, Balakrishnan K, Weigel BJ,
Highsmith WE Jr, Niu Z, Schimmenti LA. Identification of aggressive
Gardner syndrome phenotype associated with a de novo APC variant,
c.4666dup. Cold Spring Harb Mol Case Stud 2019; 5(2): a003640.
doi:10.1101/mcs.a003640. PMID: 30696621; PMCID: PMC6549566
MissingFormLabel
- 170 Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis 2017; 23: 697-708
- 171 Erkmen K, Al-Mefty O, Adada B. Tumors of the skull base. In: Oncology of CNS Tumors Philadelphia: WB Saunders; 2010: 279-307
- 172 Heller AJ, DiNardo LJ, Massey D. Fibrous dysplasia, chondrosarcoma, and McCune-Albright syndrome. Am J Otolaryngol – Head Neck Med Surg 2001; 22: 297-301
- 173 Kos M, Luczak K, Godzinski J. et al. Treatment of monostotic fibrous dysplasia with pamidronate. J Cranio-Maxillofacial Surg 2004; 32: 10-15
- 174 Collins MT, Kushner H, Reynolds JC. et al. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res 2005; 20: 219-226
- 175 Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: Age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int 2008; 19: 57-63
- 176 Devogelaer JP. New uses of bisphosphonates: Osteogenesis imperfecta. Curr Opin Pharmacol 2002; 2: 748-753
- 177 Schreiber A, Villaret AB, Maroldi R. et al. Fibrous dysplasia of the sinonasal tract and adjacent skull base. Curr Opin Otolaryngol Head Neck Surg 2012; 20: 45-52
- 178 Amit M, Collins MT, FitzGibbon EJ. et al. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia – a meta-analysis. PLoS One 2011; 6
- 179 McCune D. Osteitis fibrosa cystica: The case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child 1936; 52: 743-744
- 180 Leet AI, Chebli C, Kushner H. et al. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res 2004; 19: 571-577
- 181 Boyce AM, Brewer C, Deklotz TR. et al. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol – Head Neck Surg 2018; 144: 102-107
- 182 Deklotz TR, Kim HJ, Kelly M. et al. Sinonasal disease in polyostotic fibrous dysplasia and McCune-Albright Syndrome. Laryngoscope 2013; 123: 823-828
- 183 Boyce AM, Glover M, Kelly MH. et al. Optic neuropathy in McCune-Albright syndrome: Effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab 2013; 98
- 184 Plotkin H, Rauch F, Zeitlin L. et al. Effect of Pamidronate Treatment in Children with Polyostotic Fibrous Dysplasia of Bone. J Clin Endocrinol Metab 2003; 88: 4569-4575
- 185 DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 2007; 4: 440-445
- 186 Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis 2008; (03) 12 PMID: 18489744 PMCID: PMC2459161
- 187 Feldman MD, Kelly M, Rao VM. et al. Fibrous dysplasia of the paranasal sinuses. Otolaryngol Neck Surg 1986; 95: 222-225
- 188 Ikeda K, Suzuki H, Oshima T. et al. Endonasal endoscopic management in fibrous dysplasia of the paranasal sinuses. Am J Otolaryngol – Head Neck Med Surg 1997; 18: 415-418
- 189 Uzun C, Adali MK, Koten M. et al. McCune-Albright syndrome with fibrous dysplasia of the paranasal sinuses. Rhinology 1999; 37: 122-124
- 190 Lee JS, Fitzgibbon E, Butman JA. et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 2002; 347: 1670-1676
- 191 Wenig BM, Heffner DK. Respiratory epithelial adenomatoid hamartomas of the sinonasal tract and nasopharynx: A clinicopathologic study of 31 cases. Ann Otol Rhinol Laryngol 1995; 104: 639-645
- 192 Delbrouck C, Fernandez Aguilar S, Choufani G. et al. Respiratory epithelial adenomatoid hamartoma associated with nasal polyposis. Am J Otolaryngol – Head Neck Med Surg 2004; 25: 282-284
- 193 Endo R, Matsuda H, Takahashi M. et al. Respiratory epithelial adenomatoid hamartoma in the nasal cavity. Acta Otolaryngol 2002; 122: 398-400
- 194 Ingram WF, Noone MC, Gillespie MB. Respiratory epithelial adenomatoid hamartoma: A case report. Ear, Nose Throat J 2006; 85: 190-192
- 195 Athre R, Ducic Y. Frontal sinus hamartomas. Am J Otolaryngol – Head Neck Med Surg 2005; 26: 419-421
- 196 Flavin R, Russell J, Phelan E. et al. Chondro-osseous respiratory epithelial adenomatoid hamartoma of the nasal cavity: A case report. Int J Pediatr Otorhinolaryngol 2005; 69: 87-91
- 197 Fitzhugh VA, Mirani N. Respiratory epithelial adenomatoid hamartoma: A review. Head Neck Pathol 2008; 2: 203-208
- 198 Braun H, Beham A, Stammberger H. Respiratorisches epitheliales adenomatoides hamartom der nasenhaupthöhle: Fallbericht und literaturübersicht. Laryngorhinootologie 2003; 82: 416-420
- 199 Himi Y, Yoshizaki T, Sato K. et al. Respiratory epithelial adenomatoid hamartoma of the maxillary sinus. J Laryngol Otol 2002; 116: 317-318
- 200 Safi C, Li C, Tabaee A. et al. Outcomes and imaging findings of respiratory epithelial adenomatoid hamartoma: a systematic review. Int Forum Allergy Rhinol 2019; 9: 674-680
- 201 Lima NB, Jankowski R, Georgel T. et al. Respiratory adenomatoid hamartoma must be suspected on CT-scan enlargement of the olfactory clefts. Rhinology 2006; 44: 264-269
- 202 Hawley KA, Ahmed M, Sindwani R. CT findings of sinonasal respiratory epithelial adenomatoid hamartoma: A closer look at the olfactory clefts. Am J Neuroradiol 2013; 34: 1086-1090
- 203 Metselaar RM, Stel HV, Van Der Baan S. Respiratory epithelial adenomatoid hamartoma in the nasopharynx. J Laryngol Otol 2005; 119: 476-478
- 204 Hawley KA, Pabon S, Hoschar AP. et al. The presentation and clinical significance of sinonasal respiratory epithelial adenomatoid hamartoma (REAH). Int Forum Allergy Rhinol 2013; 3: 248-253
- 205 Schafer DR, Thompson LDR, Smith BC. et al. Primary ameloblastoma of the sinonasal tract: A clinicopathologic study of 24 cases. Cancer 1998; 82: 667-674
- 206 Shahidi S, Bronoosh P, Daneshbod Y. Follicular ameloblastoma presenting as a sinonasal tumor. Iran Red Crescent Med J 2012; 14: 113-116
- 207 Hertog D, van der Waal I. Ameloblastoma of the jaws: A critical reappraisal based on a 40-years single institution experience. Oral Oncol 2010; 46: 61-64
- 208 Barrena BG, Phillips BJ, Moran KJ. et al. Sinonasal Ameloblastoma. Head Neck Pathol 2019; 13: 247-250
- 209
Golbin DA, Ektova AP, Demin MO, Lasunin N, Cherekaev VA. Nasal
Chondromesenchymal Hamartoma with Skull Base and Orbital
Involvement: Case Presentation. Cureus 2018; 10(6): e2892.
doi:10.7759/cureus.2892. PMID: 30167348; PMCID: PMC6112910
MissingFormLabel
- 210 McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: An upper respiratory tract analogue of the chest wall mesenchymal hamartoma. Am J Surg Pathol 1998; 22: 425-433
- 211 Ozolek JA, Carrau R, Barnes EL. et al. Nasal chondromesenchymal hamartoma in older children and adults: Series and immunohistochemical analysis. Arch Pathol Lab Med 2005; 129: 1444-1450
- 212 Chan TY. World Health Organization classification of tumours: Pathology & genetics of tumours of the urinary system and male genital organs. Urology 2005; 65: 214-215
- 213 Seibert RW, Seibert JJ, Jimenez JF. et al. Nasopharyngeal brain heterotopia – a cause of upper airway obstruction in infancy. Laryngoscope 1984; 94: 818-819
- 214 Kanjanawasee D, Chaowanapanja P, Keelawat S. et al. Sphenoid Sinus Cholesteatoma – Complications and Skull Base Osteomyelitis: Case Report and Review of Literature. Clin Med Insights Case Reports 2019; 12
- 215 Hansen S, Sørensen CH, Stage J. et al. Massive cholesteatoma of the frontal sinus: Case report and review of the literature. Auris Nasus Larynx 2007; 34: 387-392
- 216
Gey A, Plontke SK, Scheller C, Kösling S, Fathke C, Glien A. Seltene
Diagnose einer knochendestruierenden Läsion der Keilbeinhöhle
[Rare diagnosis of a bone-destructive lesion of the sphenoid sinus].
HNO. 2020 Dec 14. German. doi:10.1007/s00106-020-00974-2.
Epub ahead of print. PMID: 33315128
MissingFormLabel
- 217 Ansa B, Goodman M, Ward K. et al. Paranasal sinus squamous cell carcinoma incidence and survival based on surveillance, epidemiology, and end results data, 1973 to 2009. Cancer 2013; 119: 2602-2610
- 218 Turner JH, Reh DD. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2012; 34: 877-885
- 219 Sanghvi S, Khan MN, Patel NR. et al. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2014; 124: 76-83
- 220 Youlden DR, Cramb SM, Peters S. et al. International comparisons of the incidence and mortality of sinonasal cancer. Cancer Epidemiol 2013; 37: 770-779
- 221 Hayes RB, Kardaun JWPF, De Bruyn A. Tobacco use and sinonasal cancer: A case-control study. Br J Cancer 1987; 56: 843-846
- 222 Brinton LA, Blot WJ, Becker JA. et al. A case-control study of cancers of the nasal cavity and paranasal sinuses. Am J Epidemiol 1984; 119: 896-906
- 223 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian Papilloma (Malignant Transformation): A Clinicopathologic and Immunophenotypic Study of 20 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2014; 8: 269-286
- 224 Dulguerov P, Jacobsen M, Allal A. et al. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer 2001; 92: 3012-3029
- 225 Thorup C, Sebbesen L, Danø H. et al. Carcinoma of the nasal cavity and paranasal sinuses in Denmark 1995-2004. Acta Oncol (Madr) 2010; 49: 389-394
- 226 El-Mofty SK, Lu DW. Prevalence of high-risk human papillomavirus DNA in nonkeratinizing (cylindrical cell) carcinoma of the sinonasal tract: A distinct clinicopathologic and molecular disease entity. Am J Surg Pathol 2005; 29: 1367-1372
- 227 Bishop JA, Guo TW, Smith DF. et al. Human papillomavirus-related carcinomas of the sinonasal tract. Am J Surg Pathol 2013; 37: 185-192
- 228 Osborn DA. Nature and behavior of transitional tumors in the upper respiratory tract. Cancer 1970; 25: 50-60
- 229 Robin PE, Powell DJ, Stansbie JM. Carcinoma of the nasal cavity and paranasal sinuses: incidence and presentation of different histological types. Clin Otolaryngol Allied Sci 1979; 4: 431-456
- 230 Larque AB, Hakim S, Ordi J. et al. High-risk human papillomavirus is transcriptionally active in a subset of sinonasal squamous cell carcinomas. Mod Pathol 2014; 27: 343-351
- 231 Mirghani H, Hartl D, Mortuaire G. et al. Nodal recurrence of sinonasal cancer: Does the risk of cervical relapse justify a prophylactic neck treatment?. Oral Oncol 2013; 49: 374-380
- 232 Castelnau-Marchand P, Levy A, Moya-Plana A. et al. Sinunasales Plattenepithelkarzinom ohne klinische Lymphknotenbeteiligung: Welches Halsmanagement ist das Beste?. Strahlentherapie und Onkol 2016; 192: 537-544
- 233 Nudell J, Chiosea S, Thompson LDR. Carcinoma Ex-Schneiderian
Papilloma (Malignant Transformation): A Clinicopathologic and
Immunophenotypic Study of 20 Cases Combined with a Comprehensive
Review of the Literature. Head Neck Pathol 2014; 8: 269–286
MissingFormLabel
- 234 Furuta A, Kudo M, Kanai KI. et al. Typical carcinoid tumor arising in the nose and paranasal sinuses – Case report. Auris Nasus Larynx 2010; 37: 381-385
- 235 Guan M, Li Y, Shi ZG. et al. Sarcomatoid carcinoma involving the nasal cavity and paranasal sinus: A rare and highly progressive tumor. Int J Clin Exp Pathol 2014; 7: 4489-4492
- 236 Thompson LDR, Wieneke JA, Miettinen M. et al. Spindle cell (sarcomatoid) carcinomas of the larynx: A clinicopathologic study of 187 cases. Am J Surg Pathol 2002; 26: 153-170
- 237 Lewis JE, Olsen KD, Sebo TJ. Spindle cell carcinoma of the larynx: Review of 26 cases including DNA content and immunohistochemistry. Hum Pathol 1997; 28: 664-673
- 238 Jeng YM, Sung MT, Fang CL. et al. Sinonasal undifferentiated carcinoma and nasopharyngeal-type undifferentiated carcinoma: Two clinically, biologically, and histopathologically distinct entities. Am J Surg Pathol 2002; 26: 371-376
- 239 Leung SuetYi, Yuen SiuTsan, Chung LapPing. et al. Epstein-Barr virus is present in a wide histological spectrum of sinonasal carcinomas. Am J Surg Pathol 1995; 19: 994-1001
- 240
Takakura H, Tachino H, Fujisaka M, Nakajima T, Yamagishi K, Ishida M,
Shojaku H. Lymphoepithelial carcinoma of the maxillary sinus: A case
report and review of the literature. Medicine (Baltimore) 2018;
97(28): e11371. doi:10.1097/MD.0000000000011371. PMID:
29995775; PMCID: PMC6076030
MissingFormLabel
- 241 Frierson HF, Mills SE, Fechner RE. et al. Sinonasal undifferentiated carcinoma. An aggressive neoplasm derived from Schneiderian epithelium and distinct from olfactory neuroblastoma. Am J Surg Pathol 1986; 10: 771-779
- 242 Cerilli LA, Holst VA, Brandwein MS. et al. Sinonasal undifferentiated carcinoma: Immunohistochemical profile and lack of EBV association. Am J Surg Pathol 2001; 25: 156-163
- 243
Abdelmeguid AS, Bell D, Hanna EY. Sinonasal Undifferentiated
Carcinoma. Curr Oncol Rep 2019; 21(3): 26. doi:10.1007/s11912-
019-0776-4. PMID: 30806835
MissingFormLabel
- 244 Musy PY, Reibel JF, Levine PA. Sinonasal undifferentiated carcinoma; The search for a better outcome. Laryngoscope 2002; 112: 1450-1455
- 245 Schröck A, Göke F, van Bremen T. et al. Maligne erkrankungen des sinunasaltrakts. Eine monoinstitutionelle erfahrung von 1996 bis 2010. HNO 2012; 60: 1041-1046
- 246 Rosenthal DI, Barker JL, El-Naggar AK. et al. Sinonasal malignancies with neuroendocrine differentiation: Patterns of failure according to histologic phenotype. Cancer 2004; 101: 2567-2573
- 247 Bell D, Hanna EY. Sinonasal undifferentiated carcinoma: Morphological heterogeneity, diagnosis, management and biological markers. Expert Rev Anticancer Ther 2013; 13: 285-296
- 248 Chambers KJ, Lehmann AE, Remenschneider A. et al. Incidence and survival patterns of sinonasal undifferentiated carcinoma in the United States. J Neurol Surgery, Part B Skull Base 2015; 76: 94-100
- 249 Kuan EC, Arshi A, Mallen-St Clair J, Tajudeen BA, Abemayor E, St John
MA. Significance of Tumor Stage in Sinonasal Undifferentiated
Carcinoma Survival: A Population-Based Analysis. Otolaryngol Head
Neck Surg 2016; 154(4): 667–673. doi:10.1177/01945998
16629649. Epub 2016 Feb 23. PMID: 26908559
MissingFormLabel
- 250 Takahashi Y, Lee J, Pickering C. et al. Human epidermal growth factor receptor 2/neu as a novel therapeutic target in sinonasal undifferentiated carcinoma. Head Neck 2016; 38: E1926-E1934
- 251 Sanchez-Casis G, Devine KD, Weiland LH. Nasal adenocarcinomas that closely simulate colonic carcinomas. Cancer 1971; 28: 714-720
- 252 Barnes L. Intestinal-type adenocarcinoma of the nasal cavity and paranasal sinuses. Am J Surg Pathol 1986; 10: 192-202
- 253 Szablewski V, Solassol J, Poizat F. et al. EGFR expression and KRAS and BRAF mutational status in intestinal-type sinonasal adenocarcinoma. Int J Mol Sci 2013; 14: 5170-5181
- 254 Saber AT, Nielsen LR, Dictor M. et al. K-ras mutations in sinonasal adenocarcinomas in patients occupationally exposed to wood or leather dust. Cancer Lett 1998; 126: 59-65
- 255 Franchi A, Innocenti DRD, Palomba A. et al. Low prevalence of K-RAS, EGF-R and BRAF mutations in sinonasal adenocarcinomas. Implications for anti-EGFR treatments. Pathol Oncol Res 2014; 20: 571-579
- 256 Projetti F, Durand K, Chaunavel A. et al. Epidermal growth factor receptor expression and KRAS and BRAF mutations: Study of 39 sinonasal intestinal-type adenocarcinomas. Hum Pathol 2013; 44: 2116-2125
- 257 Ferrari M, Bossi P, Mattavelli D, Ardighieri L, Nicolai P. Management of
sinonasal adenocarcinomas with anterior skull base extension. J
Neurooncol 2020;150(3): 405–417. doi:10.1007/s11060-019-
03385-8. Epub 2020 Jan 3. PMID: 31897925
MissingFormLabel
- 258 Hoffmann TK, El Hindy N, Müller OM. et al. Vascularised local and free flaps in anterior skull base reconstruction. Eur Arch Oto-Rhino-Laryngology 2013; 270: 899-907
- 259 Hoeben A, van de Winkel L, Hoebers F. et al. Intestinal-type sinonasal adenocarcinomas: The road to molecular diagnosis and personalized treatment. Head Neck 2016; 38: 1564-1570
- 260 Turri-Zanoni M, Battaglia P, Lambertoni A. et al. Treatment strategies for primary early-stage sinonasal adenocarcinoma: A retrospective bi-institutional case-control study. J Surg Oncol 2015; 112: 561-567
- 261 Licitra L, Locati LD, Cavina R. et al. Primary chemotherapy followed by anterior craniofacial resection and radiotherapy for paranasal cancer. Ann Oncol 2003; 14: 367-372
- 262 Jo VY, Mills SE, Cathro HP. et al. Low-grade sinonasal adenocarcinomas: The association with and distinction from respiratory epithelial adenomatoid hamartomas and other glandular lesions. Am J Surg Pathol 2009; 33: 401-408
- 263 Neto AG, Pineda-Daboin K, Luna MA. Sinonasal tract seromucous adenocarcinomas: A report of 12 cases. Ann Diagn Pathol 2003; 7: 154-159
- 264 Heffner DK, Hyams VJ, Hauck KW. et al. Low-grade adenocarcinoma of the nasal cavity and paranasal sinuses. Cancer 1982; 50: 312-322
- 265 Stelow EB, Jo VY, Mills SE. et al. A histologic and immunohistochemical study describing the diversity of tumors classified as sinonasal high-grade nonintestinal adenocarcinomas. Am J Surg Pathol 2011; 35: 971-980
- 266 Bignami M, Lepera D, Volpi L. et al. Sinonasal Non-Intestinal-Type Adenocarcinoma: A Retrospective Review of 22 Patients. World Neurosurg 2018; 120: e962-e969
- 267 Shay A, Ganti A, Raman A. et al. Survival in low-grade and high-grade sinonasal adenocarcinoma: A national cancer database analysis. Laryngoscope 2020; 130: E1-E10
- 268 Shen T, Shi Q, Velosa C. et al. Sinonasal renal cell-like adenocarcinomas: Robust carbonic anhydrase expression. Hum Pathol 2015; 46: 1598-1606
- 269 Heffner DK. Sinonasal and laryngeal salivary gland lesions. In: Surgical Pathology of the Salivary Glands. Philadelphia: WB Saunders; 1991: 544-559
- 270
Akbaba S, Ahmed D, Mock A, Held T, Bahadir S, Lang K, Syed M,
Hoerner-Rieber J, Forster T, Federspil P, Herfarth K, Plinkert P, Debus
J, Adeberg S. Treatment Outcome of 227 Patients with Sinonasal Adenoid
Cystic Carcinoma (ACC) after Intensity Modulated Radiotherapy
and Active Raster-Scanning Carbon Ion Boost: A 10-Year Single-Center
Experience. Cancers (Basel) 2019; 111(11): 1705–417.
doi:10.3390/cancers11111705. PMID: 31683896; PMCID:
PMC6895865
MissingFormLabel
- 271 Spiro RH, Huvos AG, Strong EW. Adenoid cystic carcinoma of salivary origin. A clinicopathologic study of 242 cases. Am J Surg 1974; 128: 512-520
- 272 Spiro RH. Salivary neoplasms: Overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8: 177-184
- 273 Michel G, Joubert M, Delemazure AS. et al. Adenoid cystic carcinoma of the paranasal sinuses: Retrospective series and review of the literature. Eur Ann Otorhinolaryngol Head Neck Dis 2013; 130: 257-262
- 274 Kim GE, Park HC, Keum KC. et al. Adenoid cystic carcinoma of the maxillary antrum. Am J Otolaryngol – Head Neck Med Surg 1999; 20: 77-84
- 275 Chen AM, Bucci MK, Weinberg V. et al. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: Prognostic features of recurrence. Int J Radiat Oncol Biol Phys 2006; 66: 152-159
- 276 Terhaard CHJ, Lubsen H, Rasch CRN. et al. The role of radiotherapy in the treatment of malignant salivary gland tumors. Int J Radiat Oncol Biol Phys 2005; 61: 103-111
- 277 Garden AS, Weber RS, Ang KK. et al. Postoperative radiation therapy for malignant tumors of minor salivary glands. Outcome and patterns of failure. Cancer 1994; 73: 2563-2569
- 278 Chen AM, Granchi PJ, Garcia J. et al. Local-regional recurrence after surgery without postoperative irradiation for carcinomas of the major salivary glands: Implications for adjuvant therapy. Int J Radiat Oncol Biol Phys 2007; 67: 982-987
- 279 Mendenhall WM, Morris CG, Amdur RJ. et al. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck 2004; 26: 154-162
- 280 Terhaard CHJ, Lubsen H, Van Der Tweel I. et al. Salivary gland carcinoma: Independent prognostic factors for locoregional control, distant metastases, and overall survival: Results of the Dutch Head and Neck Oncology Cooperative Group. Head Neck 2004; 26: 681-693
- 281 Katz TS, Mendenhall WM, Morris CG. et al. Malignant tumors of the nasal cavity and paranasal sinuses. Head Neck 2002; 24: 821-829
- 282 Laramore GE, Krall JM, Griffin TW. et al. Neutron versus photon irradiation for unresectable salivary gland tumors: Final report of an RTOG-MRC randomized clinical trial. Int J Radiat Oncol Biol Phys 1993; 27: 235-240
- 283 Douglas JG, Koh WJ, Austin-Seymour M. et al. Treatment of salivary gland neoplasms with fast neutron radiotherapy. Arch Otolaryngol - Head Neck Surg 2003; 129: 944-948
- 284 Stannard C, Vernimmen F, Carrara H. et al. Malignant salivary gland tumours: Can fast neutron therapy results point the way to carbon ion therapy?. Radiother Oncol 2013; 109: 262-268
- 285 Huber PE, Debus J, Latz D. et al. Radiotherapy for advanced adenoid cystic carcinoma: Neutrons, photons or mixed beam?. Radiother Oncol 2001; 59: 161-167
- 286 Pommier P, Liebsch NJ, Deschler DG. et al. Proton beam radiation therapy for skull base adenoid cystic carcinoma. Arch Otolaryngol - Head Neck Surg 2006; 132: 1242-1249
- 287 Patel NR, Sanghvi S, Khan MN. et al. Demografic trends and disease-specific survival in salivary acinic cell carcinoma: An analysis of 1129 cases. Laryngoscope 2014; 124: 172-178
- 288 Haerle SK, Gullane PJ, Witterick IJ. et al. Sinonasal Carcinomas. Epidemiology, Pathology, and Management. Neurosurg Clin N Am 2013; 24: 39-49
- 289 Neto AG, Pineda-Daboin K, Spencer ML. et al. Sinonasal acinic cell carcinoma: A clinicopathologic study of four cases. Head Neck 2005; 27: 603-607
- 290 Wong A, Leong JL, Ho B. Primary acinic cell carcinoma of the ethmoid sinus. Ear, Nose Throat J 2010; 89
- 291 Biron VL, Lentsch EJ, Gerry DR. et al. Case-control analysis of survival outcomes in sinonasal acinic cell carcinoma. Int Forum Allergy Rhinol 2014; 4: 507-511
- 292 Ellis MA, Graboyes EM, Day TA. et al. Prognostic factors and occult nodal disease in mucoepidermoid carcinoma of the oral cavity and oropharynx: An analysis of the National Cancer Database. Oral Oncol 2017; 72: 174-178
- 293
Auger SR,
Patel T,
Ganti A.
et al. Effect of margin status and pathological grade in treatment of sinonasal
mucoepidermoid carcinoma. Laryngoscope. 2020
MissingFormLabel
- 294 Bhattacharyya N. Survival and staging characteristics for non-squamous cell malignancies of the maxillary sinus. Arch Otolaryngol - Head Neck Surg 2003; 129: 334-337
- 295 Wolfish EB, Nelson BL, Thompson LDR. Sinonasal Tract Mucoepidermoid Carcinoma: A Clinicopathologic and Immunophenotypic Study of 19 Cases Combined with a Comprehensive Review of the Literature. Head Neck Pathol 2012; 6: 191-207
- 296 Lee YS, Ha SM, Paik SW. et al. Epithelial-myoepithelial carcinoma originating from a minor salivary gland in the nasal septum: A case report and literature review. Medicine (Baltimore) 2020; 99: e19072
- 297 Gore MR. Epithelial-myoepithelial carcinoma: A population-based survival analysis. BMC Ear, Nose Throat Disord 2018; 18
- 298 Morresi-Hauf AT, Reu S, Fertl A. Epithelial-myoepitheliales Karzinom der Trachea: Fallbericht und Literaturübersicht. Pathologe 2013; 34: 56-64
- 299 Roh JL, Chang HR, Choi SH. et al. Clinical utility of 18F-FDG PET for patients with salivary gland malignancies. J Nucl Med 2007; 48: 240-246
- 300 Kim CH, Jeong JS, Kim SR. et al. Endobronchial epithelial-myoepithelial carcinoma of the lung. Thorax 2018; 73: 593-594
- 301 Kim SH, Park SE, Bae HG. et al. Epithelial-myoepithelial carcinoma of the nasopharynx: A case report and review of the literature. Oncol Lett 2015; 10: 927-930
- 302 Fried D, Zanation AM, Huang B. et al. Management of nonesthesioneuroblastoma sinonasal malignancies with neuroendocrine differentiation. Laryngoscope 2012; 122: 2210-2215
- 303 Hong SL, Kim SD, Roh HJ. et al. The sphenoid sinus: An unusual presentation of a typical carcinoid tumor. J Craniofac Surg 2014; 25: e483-e485
- 304 Konukiewitz B, Agaimy A, Weichert W. et al. Neuroendokrine Neoplasien der Kopf-Hals-Region. Pathologe 2018; 39: 27-34
- 305 Ferlito A, Silver CE, Bradford CR. et al. Neuroendocrine neoplasms of the larynx: An overview. Head Neck 2009; 31: 1634-1646
- 306 Lee DH, Cho HH, Cho YB. Typical carcinoid tumor of the nasal cavity. Auris Nasus Larynx 2007; 34: 537-539
- 307 Patel TD, Carniol ET, Vázquez A. et al. Sinonasal fibrosarcoma: Analysis of the Surveillance, Epidemiology, and End Results database. Int Forum Allergy Rhinol 2016; 6: 201-205
- 308 Lartigau E, Lusinchi A, Schwaab G. Sarcomas of nasal cavity and paranasal sinuses: Chondrosarcoma, osteosarcoma and fibrosarcoma. J Laryngol Otol 1994; 108: 947-953
- 309 Koshy M, Rich SE, Mohiuddin MM. Improved Survival With Radiation Therapy in High-Grade Soft Tissue Sarcomas of the Extremities: A SEER Analysis. Int J Radiat Oncol Biol Phys 2010; 77: 203-209
- 310 Wang CP, Chang YL, Ting LL. et al. Malignant fibrous histiocytoma of the sinonasal tract. Head Neck 2009; 31: 85-93
- 311 Szablewski V, Neuville A, Terrier P. et al. Adult sinonasal soft tissue sarcoma: Analysis of 48 cases from the French Sarcoma Group database. Laryngoscope 2015; 125: 615-623
- 312 Gerrand CH, Bell RS, Wunder JS. et al. The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer 2003; 97: 485-492
- 313 Kwok MMK, Lee S, Hosking P. Leiomyosarcoma: A rare sinonasal malignancy. BMJ Case Rep 2018; 2018
- 314 Tanaka H, Westesson PL, Wilbur DC. Leiomyosarcoma of the maxillary sinus: CT and MRI findings. Br J Radiol 1998; 71: 221-224
- 315 Dobben GD. Leiomyosarcoma of the Nasopharynx. AMA Arch Otolaryngol 1958; 68: 211-213
- 316 Sanghvi S, Misra P, Patel NR. et al. Incidence trends and long-term survival analysis of sinonasal rhabdomyosarcoma. Am J Otolaryngol - Head Neck Med Surg 2013; 34: 682-689
- 317 Wurm J, Constantinidis J, Grabenbauer GG. et al. Rhabdomyosarcomas of the nose and paranasal sinuses: Treatment results in 15 cases. Otolaryngol - Head Neck Surg 2005; 133: 42-50
- 318 Narula AA, Vallis MP, El-Silimy OE. et al. Radiation induced angiosarcomas of the nasopharynx. Eur J Surg Oncol 1986; 12: 147-152
- 319 Maddox JC, Evans HL. Angiosarcoma of skin and soft tissue: A study of forty-four cases. Cancer 1981; 48: 1907-1921
- 320 Williamson IG, Ramsden RT. Angiosarcoma of maxillary antrum—association with vinyL chloride exposure. J Laryngol Otol 1988; 102: 464-467
- 321 Nelson BL, Thompson LDR. Sinonasal tract angiosarcoma: A clinicopathologic and immunophenotypic study of 10 cases with a review of the literature. Head Neck Pathol 2007; 1: 1-12
- 322 Fukushima K, Dejima K, Koike S. et al. A case of angiosarcoma of the nasal cavity successfully treated with recombinant interleukin-2. Otolaryngol - Head Neck Surg 2006; 134: 886-887
- 323 Treviño-González JL, Santos-Lartigue R, González-Andrade B. et al. Angiosarcoma of the nasal cavity: A case report. Cases J 2009; 2
- 324 Es-Sbissi F, Nitassi S, Boulaadas M. et al. Sinonasal angiosarcoma. Eur Ann Otorhinolaryngol Head Neck Dis 2015; 132: 161-163
- 325 Agaimy A, Hartmann A. Kopf-Hals-Tumoren: Neues aus der WHO-Klassifikation 2017. Pathologe 2018; 39
- 326 Agaimy A, Haller F, Hartmann A. Sinunasale Tumoren: Neues aus der WHO mit besonderem Fokus auf neue mesenchymale Entitäten. Pathologe 2018; 39: 18-26
- 327 Lewis JT, Oliveira AM, Nascimento AG. et al. Low-grade sinonasal sarcoma with neural and myogenic features: A clinicopathologic analysis of 28 cases. Am J Surg Pathol 2012; 36: 517-525
- 328 Rooper LM, Huang SC, Antonescu CR. et al. Biphenotypic sinonasal sarcoma: An expanded immunoprofile including consistent nuclear β-catenin positivity and absence of SOX10 expression. Hum Pathol 2016; 55: 44-50
- 329 Carter CS, East EG, McHugh JB. Biphenotypic sinonasal sarcoma: A review and update. In: Archives of Pathology and Laboratory Medicine. College of American Pathologists 2018; 1196-1201
- 330 Fritchie KJ, Jin L, Wang X. et al. Fusion gene profile of biphenotypic sinonasal sarcoma: an analysis of 44 cases. Histopathology 2016; 69: 930-936
- 331 Ducatman BS, Scheithauer BW, Piepgras DG. et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57: 2006-2021
- 332 Rodriguez FJ, Folpe AL, Giannini C. et al. Pathology of peripheral nerve sheath tumors: Diagnostic overview and update on selected diagnostic problems. Acta Neuropathol 2012; 123: 295-319
- 333 James AW, Shurell E, Singh A. et al. Malignant Peripheral Nerve Sheath Tumor. Surg Oncol Clin N Am 2016; 25: 789-802
- 334 Bradtmöller M, Hartmann C, Zietsch J. et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One 2012; 7: e47595
- 335 Kar M, Suryanarayana Deo SV, Shukla NK. et al. Malignant peripheral nerve sheath tumors (MPNST) - Clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol 2006; 4: 55
- 336 Jo VY, Fletcher CDM. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014; 46: 95-104
- 337 Van De Rijn M, Barr FG, Xiong QBin. et al. Radiation-associated synovial sarcoma. Hum Pathol 1997; 28: 1325-1328
- 338 Deraedt K, Debiec-Rychter M, Sciot R. Radiation-associated synovial sarcoma of the lung following radiotherapy for pulmonary metastasis of Wilms’ tumour [7]. Histopathology 2006; 48: 473-475
- 339 Egger JF, Coindre JM, Benhattar J. et al. Radiation-associated synovial sarcoma: Clinicopathologic and molecular analysis of two cases. Mod Pathol 2002; 15: 998-1004
- 340 Sherman KL, Wayne JD, Chung J. et al. Assessment of multimodality therapy use for extremity sarcoma in the United States. J Surg Oncol 2014; 109: 395-404
- 341 Vlenterie M, Jones RL, Van Der Graaf WTA. Synovial sarcoma diagnosis and management in the era of targeted therapies. Curr Opin Oncol 2015; 27: 316-322
- 342 De Bree E, Zoras O, Hunt JL. et al. Desmoid tumors of the head and neck: A therapeutic challenge. Head Neck 2014; 36: 1517-1526
- 343 Orphanet: Desmoid type fibromatosis. Im Internet: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=DE&data_id=8665&Disease_Disease_Search_diseaseType=ORPHA&Disease_Disease_Search_diseaseGroup=873&Disease(s)/groupofdiseases=Desmoid-type-fibromatosis&title=Desmoid-type-fibromatosis&search=Dise
MissingFormLabel
- 344 Colombo C, Foo WC, Whiting D. et al. FAP-related desmoid tumors: A series of 44 patients evaluated in a cancer referral center. Histol Histopathol 2012; 27: 641-649
- 345 Coffin CM, Hornick JL, Zhou H. et al. Gardner fibroma: A clinicopathologic and immunohistochemical analysis of 45 patients with 57 fibromas. Am J Surg Pathol 2007; 31: 410-416
- 346 Emori M, Kaya M, Mitsuhashi T. et al. Desmoid tumor-associated pain is dependent on mast cell expression of cyclooxygenase-2. Diagn Pathol 2014; 9
- 347 Kasper B, Baumgarten C, Garcia J. et al. An update on the management of sporadic desmoid-type fibromatosis: A European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol 2017; 28: 2399-2408
- 348 Bonvalot S, Eldweny H, Haddad V. et al. Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 2008; 34: 462-468
- 349 Penel N, Chibon F, Salas S. Adult desmoid tumors: Biology, management and ongoing trials. Curr Opin Oncol 2017; 29: 268-274
- 350 Huang PW, Tzen CY. Prognostic factors in desmoid-type fibromatosis: A clinicopathological and immunohistochemical analysis of 46 cases. Pathology 2010; 42: 147-150
- 351 Granter SR, Badizadegan K, Fletcher CDM. Myofibromatosis in adults, glomangiopericytoma, and myopericytoma: A spectrum of tumors showing perivascular myoid differentiation. Am J Surg Pathol 1998; 22: 513-525
- 352 Asimakopoulos P, Syed MI, Andrews T. et al. Sinonasal glomangiopericytoma: Is anything new?. Ear, Nose Throat J 2016; 95: E1
- 353 Gengler C, Guillou L. Solitary fibrous tumour and haemangiopericytoma: Evolution of a concept. Histopathology 2006; 48: 63-74
- 354 Thompson LDR, Miettinen M, Wenig BM. Sinonasal-type hemangiopericytoma: A clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol 2003; 27: 737-749
- 355 Catalano PJ, Brandwein M, Shah DK. et al. Sinonasal hemangiopericytomas: A clinicopathologic and immunohistochemical study of seven cases. Head Neck 1996; 18: 42-53
- 356 Billings KR, Fu YS, Calcaterra TC. et al. Hemangiopericytoma of the head and neck. Am J Otolaryngol - Head Neck Med Surg 2000; 21: 238-243
- 357 Carew JF, Singh B, Kraus DH. Hemangiopericytoma of the head and neck. Laryngoscope 1999; 109: 1409-1411
- 358 Weber W, Henkes H, Metz KA. et al. Haemangiopericytoma of the nasal cavity. Neuroradiology 2001; 43: 183-186
- 359 Witkin GB, Rosai J. Solitary fibrous tumor of the upper respiratory tract: A report of six cases. Am J Surg Pathol 1991; 15: 842-848
- 360 Batsakis JG, El-Naggar AK, Hybels RD. Pathology consultation solitary fibrous tumor. Ann Otol Rhinol Laryngol 1993; 102: 74-76
- 361 Künzel J, Hainz M, Ziebart T. et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Oto-Rhino-Laryngology 2016; 273: 1589-1598
- 362
Demicco EG,
Park MS,
Araujo DM.
et al. Solitary fibrous tumor: A clinicopathological study of 110 cases and proposed
risk assessment model. Mod Pathol 2012; 25: 1298-1306
MissingFormLabel
- 363 Xue Y, Chai G, Xiao F. et al. Post-operative radiotherapy for the treatment of malignant solitary fibrous tumor of the nasal and paranasal area. Jpn J Clin Oncol 2014; 44: 926-931
- 364 Piccaluga PP, De Falco G, Kustagi M. et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood 2011; 117: 3596-3608
- 365 Chi AC, Weathers DR, Folpe AL. et al. Epithelioid hemangioendothelioma of the oral cavity: Report of two cases and review of the literature. Oral Surgery, Oral Med Oral Pathol Oral Radiol Endodontology 2005; 100: 717-724
- 366 Errani C, Zhang L, Sung YS. et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosom Cancer 2011; 50: 644-653
- 367 Flucke U, Vogels RJC, de Saint Aubain Somerhausen N. et al. Epithelioid Hemangioendothelioma: Clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol 2014; 9
- 368 Bruder E, Alaggio R, Kozakewich HPW. et al. Vascular and perivascular lesions of skin and soft tissues in children and adolescents. Pediatr Dev Pathol 2012; 15: 26-61
- 369 Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma a vascular tumor often mistaken for a carcinoma. Cancer 1982; 50: 970-981
- 370 Mentzel T, Beham A, Calonje E. et al. Epithelioid hemangioendothelioma of skin and soft tissues: Clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol 1997; 21: 363-374
- 371 Deyrup AT, Tighiouart M, Montag AG. et al. Epithelioid hemangioendothelioma of soft tissue: A proposal for risk stratification based on 49 cases. Am J Surg Pathol 2008; 32: 924-927
- 372 Beger M, Antoni C, Haas S. et al. Epitheloides Hämangioendotheliom- drei Patienten, drei Therapieoptionen. Z Gastroenterol 2006; 44: A5_04
- 373 Wong BLK, Lee VNY, Tikka T. et al. Kaposiform haemangioendothelioma of the head and neck. Crit Rev Oncol Hematol 2016; 104: 156-168
- 374 Sun ZJ, Zhang L, Zhang WF. et al. Kaposiform hemangioendothelioma involving the neck. Oral Oncol Extra 2006; 42: 60-65
- 375 Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis 2013; 8: 95
- 376 Pongpruttipan T, Sukpanichnant S, Assanasen T. et al. Extranodal NK/T-cell lymphoma, nasal type, includes cases of natural killer cell and αβ, γδ, and αβ/γδ T-cell origin: A comprehensive clinicopathologic and phenotypic study. Am J Surg Pathol 2012; 36: 481-499
- 377 Jhuang JY, Chang ST, Weng SF. et al. Extranodal natural killer/T-cell lymphoma, nasal type in Taiwan: A relatively higher frequency of T-cell lineage and poor survival for extranasal tumors. Hum Pathol 2015; 46: 313-321
- 378 De Campos-Lima PO, Gavioli R, Zhang QJ. et al. HLA-A11 epitope loss isolates of Epstein-Barr virus from a highly A11 + population. Science (80-) 1993; 260: 98-100
- 379 Lima PODC, Levitsky V, Brooks J. et al. T cell responses and virus evolution: Loss of hla all-restricted ctl epitopes in epstein-barr virus isolates from highly all-positive populations by selective mutation of anchor residues. J Exp Med 1994; 179: 1297-1305
- 381 Kim SJ, Choi JY, Hyun SH. et al. Risk stratification on the basis of Deauville score on PET-CT and the presence of Epstein-Barr virus DNA after completion of primary treatment for extranodal natural killer/T-cell lymphoma, nasal type: A multicentre, retrospective analysis. Lancet Haematol 2015; 2: e66-e74
- 381 Tse E, Kwong YL. How i treat NK/T-cell lymphomas. Blood 2013; 121: 4997-5005
- 382 Kwong YL, Kim WS, Lim ST. et al. SMILE for natural killer/T-cell lymphoma: Analysis of safety and efficacy from the Asia Lymphoma Study Group. Blood 2012; 120: 2973-2980
- 383 Alexiou C, Kau RJ, Dietzfelbinger H. et al. Extramedullary plasmacytoma: Tumor occurrence and therapeutic concepts. Cancer 1999; 85: 2305-2314
- 384 Miller FR, Lavertu P, Wanamaker JR. et al. Plasmacytomas of the head and neck. Otolaryngol - Head Neck Surg 1998; 119: 614-618
- 385 Patel TD, Vázquez A, Choudhary MM. et al. Sinonasal extramedullary plasmacytoma: A population-based incidence and survival analysis. Int Forum Allergy Rhinol 2015; 5: 862-869
- 386 Soutar R, Lucraft H, Jackson G. et al. Guidelines on the diagnosis and management of solitary plasmacytoma of bone and solitary extramedullary plasmacytoma. Br J Haematol 2004; 124: 717-726
- 387 Lorsbach RB, Hsi ED, Dogan A. et al. Plasma cell myeloma and related neoplasms. In: American Journal of Clinical Pathology 2011; 168-182
- 388 Susnerwala SS, Shanks JH, Banerjee SS. et al. Extramedullary plasmacytoma of the head and neck region: Clinicopathological correlation in 25 cases. Br J Cancer 1997; 75: 921-927
- 389 Hussong JW, Perkins SL, Schnitzer B. et al. Extramedullary plasmacytoma: A form of marginal zone cell lymphoma?. Am J Clin Pathol 1999; 111: 111-116
- 390 Boll M, Parkins E, O’Connor SJM. et al. Extramedullary plasmacytoma are characterized by a „myeloma-like“ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol 2010; 151: 525-527
- 391 Galieni P, Cavo M, Pulsoni A. et al. Clinical outcome of extramedullary plasmacytoma. Haematologica 2000; 85: 47-51
- 392 Sasaki R, Yasuda K, Abe E. et al. Multi-institutional analysis of solitary extramedullary plasmacytoma of the head and neck treated with curative radiotherapy. Int J Radiat Oncol Biol Phys 2012; 82: 626-634
- 393 Knowling MA, Harwood AR, Bergsagel DE. Comparison of extramedullary plasmacytomas with solitary and multiple plasma cell tumors of bone. J Clin Oncol 1983; 1: 255-262
- 394 Hamre M, Hedberg J, Buckley J. et al. Langerhans cell histiocytosis: An exploratory epidemiologic study of 177 cases. Med Pediatr Oncol 1997; 28: 92-97
- 395 Laman JD, Leenen PJM, Annels NE. et al. Langerhans-cell histiocytosis „insight into DC biology“. Trends Immunol 2003; 24: 190-196
- 396 Orphanet: Langerhans Zell Histiozytose. Im Internet: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=DE&Expert=389
MissingFormLabel
- 397 Kostyra K, Kostkiewicz B. Langerhans cell histiocytosis of the orbit and frontal sinus of the adult woman: A first case report in Poland. Surg Neurol Int 2019; 10: 234
- 398 Mani S, Thomas R, Mathew J. et al. Langerhan’s cell histiocytosis of sphenoid sinus causing vision loss: A case report. J Nepal Med Assoc 2019; 57: 281-284
- 399 Gündüz K, Palamar M, Parmak N. et al. Eosinophilic granuloma of the orbit: Report of two cases. J AAPOS 2007; 11: 506-508
- 400 Esmaili N, Harris GJ. Langerhans Cell Histiocytosis of the Orbit: Spectrum of Disease and Risk of Central Nervous System Sequelae in Unifocal Cases. Ophthal Plast Reconstr Surg 2016; 32: 28-34
- 401 Prayer D, Grois N, Prosch H. et al. MR imaging presentation of intracranial disease associated with langerhans cell histiocytosis. Am J Neuroradiol 2004; 25: 880-891
- 402 Allen CE, Li L, Peters TL. et al. Cell-Specific Gene Expression in Langerhans Cell Histiocytosis Lesions Reveals a Distinct Profile Compared with Epidermal Langerhans Cells. J Immunol 2010; 184: 4557-4567
- 403 Hyman DM, Diamond EL, Vibat CRT. et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015; 5: 64-71
- 404 Berres ML, Lim KPH, Peters T. et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014; 211: 669-683
- 405 Wladis EJ, Tomaszewski JE, Gausas RE. Langerhans cell histiocytosis of the orbit 10 years after involvement at other sites. Ophthal Plast Reconstr Surg 2008; 24: 142-143
- 406 Herwig MC, Wojno T, Zhang Q. et al. Langerhans Cell Histiocytosis of the Orbit: Five Clinicopathologic Cases and Review of the Literature. Surv Ophthalmol 2013; 58: 330-340
- 407 Windfuhr JP. Primitive neuroectodermal tumor of the head and neck: Incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 2004; 113: 533-543
- 408 Hafezi S, Seethala RR, Stelow EB. et al. Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone. Head Neck Pathol 2011; 5: 8-16
- 409 Specht K, Sung YS, Zhang L. et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged ewing sarcomas: Further evidence toward distinct pathologic entities. Genes Chromosom Cancer 2014; 53: 622-633
- 410 Lin JK, Liang J. Sinonasal Ewing Sarcoma: A Case Report and Literature Review. Perm J 2018; 22
- 411 Allam A, El-Husseiny G, Khafaga Y. et al. Ewing’s sarcoma of the head and neck: A retrospective analysis of 24 cases. Sarcoma 1999; 3: 11-15
- 412 Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010; 11: 184-192
- 413 Broich G, Pagliari A, Ottaviani F. Esthesioneuroblastoma: A general review of the cases published since the discovery of the tumour in 1924. Anticancer Res 1997; 17: 2683-2706
- 414 Roussel LM, Patron V, Maubert E. et al. New landmarks in endonasal surgery: from nasal bone to anterior cribriform plate including branches of anterior ethmoidal artery and nerve and terminal nerve. Int Forum Allergy Rhinol 2020; 10: 395-404
- 415 Mills SE. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 2002; 15: 264-278
- 416 Thompson LDR. Olfactory Neuroblastoma. Head Neck Pathol 2009; 3: 252-259
- 417 Kadish S, Goodman M, Wang CC. Olfactory neuroblastoma—A clinical analysis of 17 cases. Cancer 1976; 37: 1571-1576
- 418 Morita A, Ebersold MJ, Olsen KD. et al. Esthesioneuroblastoma: Prognosis and management. Neurosurgery 1993; 32: 706-715
- 419 Edge SB, Compton CC. The american joint committee on cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010; 17: 1471-1474
- 420 Norton A. Tumors of the Upper Respiratory Tract and Ear. In: Hyams V, Batsakis J, Michaels L Hrsg. Journal of Clinical Pathology 1989; 335-335
- 421 Mao L, Xia YP, Zhou YN. et al. Activation of sonic hedgehog signaling pathway in olfactory neuroblastoma. Oncology 2009; 77: 231-243
- 422 Ketcham AS, Wilkins RH, Van Buren JM. et al. A combined intracranial facial approach to the paranasal sinuses. Am J Surg 1963; 106: 698-703
- 423 Hanna E, DeMonte F, Ibrahim S. et al. Endoscopic resection of sinonasal cancers with and without craniotomy: Oncologic results. Arch Otolaryngol - Head Neck Surg 2009; 135: 1219-1224
- 424 Su SY, Bell D, Ferrarotto R. et al. Outcomes for olfactory neuroblastoma treated with induction chemotherapy. Head Neck 2017; 39: 1671-1679
- 425 Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: A meta-analysis and review. Lancet Oncol 2001; 2: 683-690
- 426 Ow TJ, Hanna EY, Roberts DB. et al. Optimization of long-term outcomes for patients with esthesioneuroblastoma. Head Neck 2014; 36: 524-530
- 427
Almutuawa DM,
Strohl MP,
Gruss C.
et al. Outcomes of sinonasal mucosal melanomas with endoscopic and open resection:
a
retrospective cohort study. J Neurooncol. 2020
MissingFormLabel
- 428 Thompson LDR, Wieneke JA, Miettinen M. Sinonasal tract and nasopharyngeal melanomas: A clinicopathologic study of 115 cases with a proposed staging system. Am J Surg Pathol 2003; 27: 594-611
- 429 Moreno MA, Roberts DB, Kupferman ME. et al. Mucosal melanoma of the nose and paranasal sinuses, a contemporary experience from the M. D. Anderson cancer center. Cancer 2010; 116: 2215-2223
- 430 Patel SG, Prasad ML, Escrig M. et al. Primary mucosal malignant melanoma of the head and neck. Head Neck 2002; 24: 247-257
- 431 Zebary A, Jangard M, Omholt K. et al. KIT, NRAS and BRAF mutations in sinonasal mucosal melanoma: A study of 56 cases. Br J Cancer 2013; 109: 559-564
- 432 Van Essen TH, Van Pelt S, Versluis M. et al. Prognostic parameters in uveal melanoma and their association with bap1 expression. Br J Ophthalmol 2014; 98: 1738-1743
- 433 Dauer EH, Lewis JE, Rohlinger AL. et al. Sinonasal melanoma: A clinicopathologic review of 61 cases. Otolaryngol - Head Neck Surg 2008; 138: 347-352
- 434 Bachar G, Kwok SL, O’Sullivan B. et al. Mucosal melanomas of the head and neck: The Princess Margaret Hospital experience. Head Neck 2008; 30: 1325-1331
- 435
Lund VJ,
Chisholm EJ,
Howard DJ.
et al. Sinonasal malignant melanoma: An analysis of 115 cases assessing outcomes of
surgery, postoperative radiotherapy and endoscopic resection. Rhinology 2012; 50:
203-210
MissingFormLabel
- 436 Thierauf J, Glück AM, Plinkert P. et al. Mucosal melanoma of the cranio-facial region: Surgical challenges and therapeutic options. Auris Nasus Larynx 2019; 46: 252-259
- 437 Gore MR, Zanation AM. Survival in sinonasal melanoma: A meta-analysis. J Neurol Surgery, Part B Skull Base 2012; 73: 157-162
- 438 Ganly I, Patel SG, Singh B. et al. Craniofacial resection for malignant melanoma of the skull base: Report of an International Collaborative Study. Arch Otolaryngol - Head Neck Surg 2006; 132: 73-78
- 439 Jarrom D, Paleri V, Kerawala C. et al. Mucosal melanoma of the upper airways tract mucosal melanoma: A systematic review with meta-analyses of treatment. Head Neck 2017; 39: 819-825
- 440 Li J, Kan H, Zhao L. et al. Immune checkpoint inhibitors in advanced or metastatic mucosal melanoma: a systematic review. Ther Adv Med Oncol 2020; 12
- 441 Misra P, Husain Q, Svider PF. et al. Management of sinonasal teratocarcinosarcoma: A systematic review. Am J Otolaryngol - Head Neck Med Surg 2014; 35: 5-11
- 442 Chao KK, Eng TY, Barnes J. et al. Sinonasal Teratocarcinosarcoma. Am J Clin Oncol Cancer Clin Trials 2004; 27: 29-32
- 443 Fukuoka K, Hirokawa M, Shimizu M. et al. Teratocarcinosarcoma of the nasal cavity. Report of a case showing favorable prognosis. Apmis 2000; 108: 553-557
- 444 Heffner DK, Hyams VJ. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses: A clinicopathologic study of 20 cases. Cancer 1984; 53: 2140-2154
- 445 Wei S, Carroll W, Lazenby A. et al. Sinonasal teratocarcinosarcoma: report of a case with review of literature and treatment outcome. Ann Diagn Pathol 2008; 12: 415-425
- 446 Vranic S, Caughron SK, Djuricic S. et al. Hamartomas, teratomas and teratocarcinosarcomas of the head and neck: Report of 3 new cases with clinico-pathologic correlation, cytogenetic analysis, and review of the literature. BMC Ear, Nose Throat Disord 2008; 8
- 447 Lim CCT, Thiagarajan A, Sim CS. et al. Craniospinal dissemination in teratocarcinosarcoma: Case report. J Neurosurg 2008; 109: 321-324
- 448 Terasaka S, Medary MB, Whiting DM. et al. Prolonged survival in a patient with sinonasal teratocarcinosarcoma with cranial extension. Case report. J Neurosurg 1998; 88: 753-756
- 449 Dicke TE, Gates GA. Malignant Teratoma of the Paranasal Sinuses: Report of a Case. Arch Otolaryngol 1970; 91: 391-394



















